Arch Iran Med. 28(12):719-722.
doi: 10.34172/aim.34740
Case Report
Pancreatic PEComa: Case Report of an Extremely Rare Tumor
Dmitry Zinovkin Conceptualization, Project administration, Software, Validation, 1, * 
Denis A. Davydov Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing, 2
Pavel G. Kisialeu Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing, 2
Diana A. Kolbik Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing, 2
Sergey L. Achinovich Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing, 3
Anna S. Portyanko Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing, 2
Md Zahidul Islam Pranjol Conceptualization, Project administration, Software, Validation, 4, *
Author information:
1Department of Pathology, Gomel State Medical University, Gomel, Belarus
2National Molecular Genetics Laboratory of Cancer Research, N.N.Alexandrov National Cancer Center of Belarus, Minsk, Belarus
3Department of Pathology, Gomel Regional Oncological Clinics, Gomel, Belarus
4School of Life Sciences, University of Sussex, Brighton, UK
Abstract
Pancreatic perivascular epithelioid cell tumors (PEComas) are rare mesenchymal neoplasms with only a few reported cases. Their non-specific clinical presentations and imaging features often lead to misdiagnosis. We report a case of a 63-year-old female with intermittent left upper quadrant pain. Imaging revealed a hypervascular mass in the pancreatic tail, initially suspected to be a neuroendocrine tumor. The patient underwent distal pancreatectomy with splenectomy. Histopathological examination showed that the tumor consisted of epithelioid and spindle cells with clear cytoplasm, a rich vascular network and low mitotic activity. Immunohistochemically, the tumor cells were positive for HMB-45, Melan-A, and smooth muscle actin, confirming the diagnosis of pancreatic PEComa. The postoperative course was uneventful. Given the uncertain malignant potential of PEComas, complete surgical excision is the preferred treatment option, with long-term follow-up recommended. This case highlights the diagnostic challenges of pancreatic PEComas and underscores the role of histopathology and immunohistochemistry in their accurate identification and management.
Keywords: Diagnostic challenges, Immunohistochemistry, Pancreatic PEComa, Rare mesenchymal tumor, Surgical resection
Copyright and License Information
© 2025 The Author(s).
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article as: Zinovkin D, Davydov DA, Kisialeu PG, Kolbik DA, Achinovich SL, Portyanko AS, et al. Pancreatic PEComa: case report of an extremely rare tumor. Arch Iran Med. 2025;28(12):719-722. doi: 10.34172/aim.34740
Introduction
Perivascular epithelioid cell tumors (PEComas) are rare mesenchymal neoplasms characterized by the presence of perivascular epithelioid cells that co-express melanocytic and smooth muscle markers. While these tumors have been reported in various anatomical locations, including the kidneys, lungs, liver, and uterus, their occurrence in the pancreas is extremely rare.1 The first documented case of a pancreatic PEComa was reported by Zamboni et al in 1996, and since then, only about 30 cases have been described.2
Pancreatic PEComas present with diverse clinical manifestations, ranging from incidental detection to symptoms related to mass effect, such as abdominal pain, weight loss, or obstructive complications.3 Due to their rarity and non-specific radiological features, pancreatic PEComas are often challenging to diagnose preoperatively. Histopathological and immunohistochemical evaluations are essential for confirmation, as tumors typically express melanocytic markers such as HMB-45 and Melan-A, in addition to smooth muscle markers like α-smooth muscle actin.3
The biological behavior of pancreatic PEComas remains uncertain; some cases exhibit benign features, while others demonstrate aggressive growth, local invasion, or metastatic potential.4 Given their unpredictable nature, surgical resection is generally considered as the primary treatment option, especially for tumors with worrisome histopathological characteristics.5
In this report, we present a case of pancreatic PEComa, detailing its clinical presentation, diagnostic findings, histopathological features, and treatment approach. This case contributes to the existing literature and emphasizes the importance of accurate diagnosis and appropriate management of these rare pancreatic neoplasms.
Case Report
A 63-year-old female patient presented with a two-year history of intermittent pain in the left upper quadrant. She had no significant past medical or surgical history. On admission, her weight was 94 kg, height 164 cm, and blood pressure 130/80 mmHg. Electrocardiography revealed sinus bradycardia (58 bpm), left axis deviation, and first-degree atrioventricular block. Laboratory tests showed mild anemia (hemoglobin 109 g/L), thrombocytosis (510 × 10⁹/L), and an elevated erythrocyte sedimentation rate (56 mm/h). Biochemical parameters were largely within normal limits, except for hyperglycemia (12.0 mmol/L) and a slightly elevated alkaline phosphatase level (136.1 U/L).
Abdominal contrast-enhanced multi-slice computed tomography revealed a hypervascular mass measuring 30 × 21 × 25 mm in the tail of the pancreas (Figure 1), suspected to be neuroendocrine neoplasm. The lesion demonstrated early arterial enhancement with persistent contrast uptake in the venous phase, without pancreatic duct dilatation or invasion of adjacent structures. There were no radiologic signs of regional lymphadenopathy or distant metastases. Given the well-circumscribed nature of the mass and its location in the pancreatic tail, the surgical team opted for upfront resection instead of preoperative endoscopic ultrasound-guided fine-needle aspiration (EUS-FNA), to both avoid procedure-related risks and obtain a complete specimen for histopathological and immunohistochemical analysis. The patient underwent a distal subtotal pancreatectomy with splenectomy.
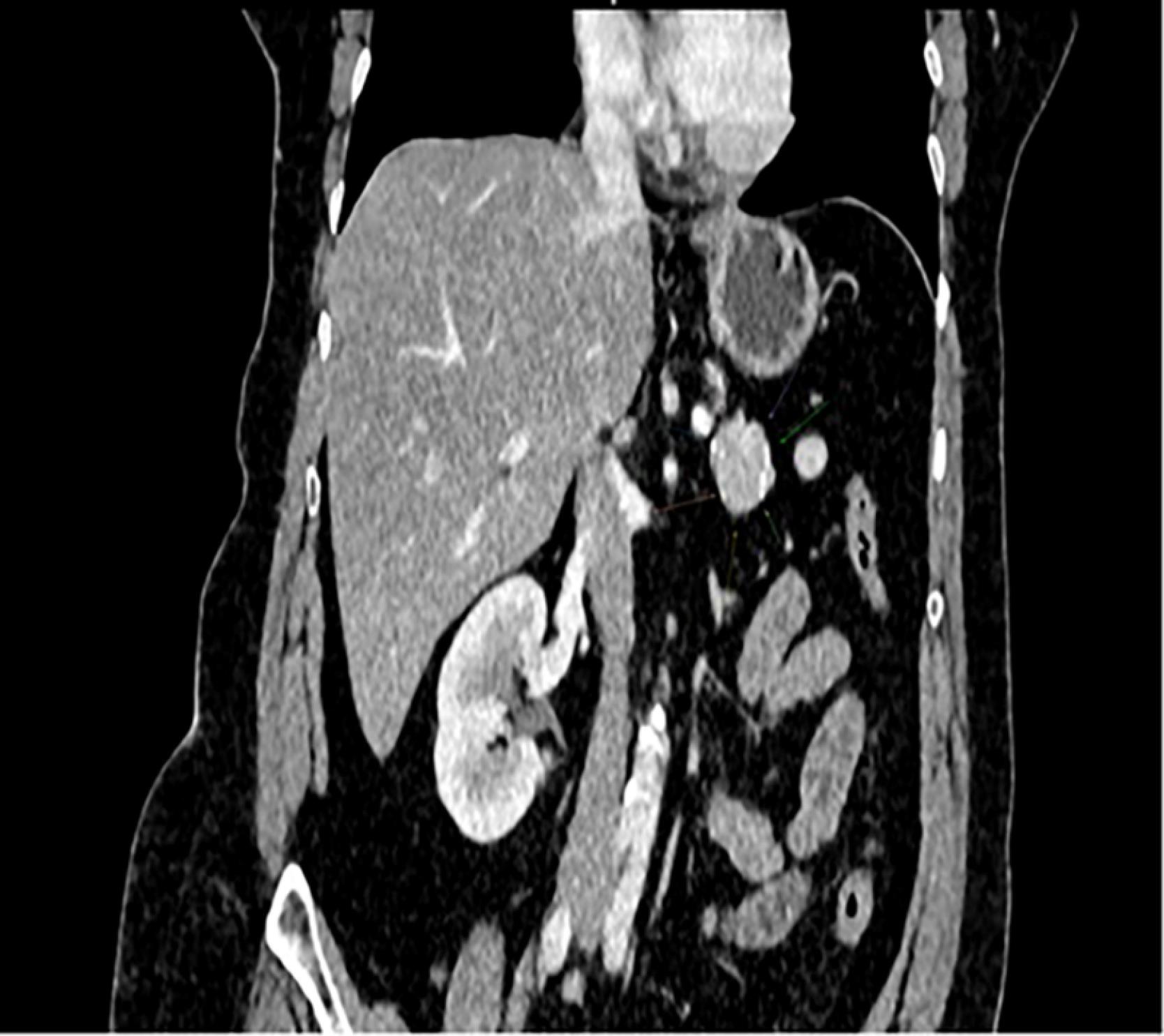
Figure 1.
Well-circumscribed solitary tumor of the pancreas. Contrast-enchanced CT scan, sagittal plane
.
Well-circumscribed solitary tumor of the pancreas. Contrast-enchanced CT scan, sagittal plane
Macroscopic examination of the resected specimen showed a well-circumscribed, solid, lobulated tumor in the pancreatic tail, measuring 30 × 21 × 25 mm, surrounded by a fibrous capsule. The lesion appeared pale brown in color. The spleen, measuring 95 × 45 × 55 mm and weighing 120 g, had an intact capsule except for a minor tear at the hilum.
Histopathologically, the tumor had a thick fibrous capsule at the periphery, separating the lesion from the adjacent pancreatic tissue. The tumor consisted of large, predominantly epithelioid cells with clear and granular cytoplasm, round nuclei without prominent nucleoli. Occasional multinucleated cells were found, as well as minor areas of spindle-cell architecture. The mitotic rate was low, and necrosis was absent (Figure 2).
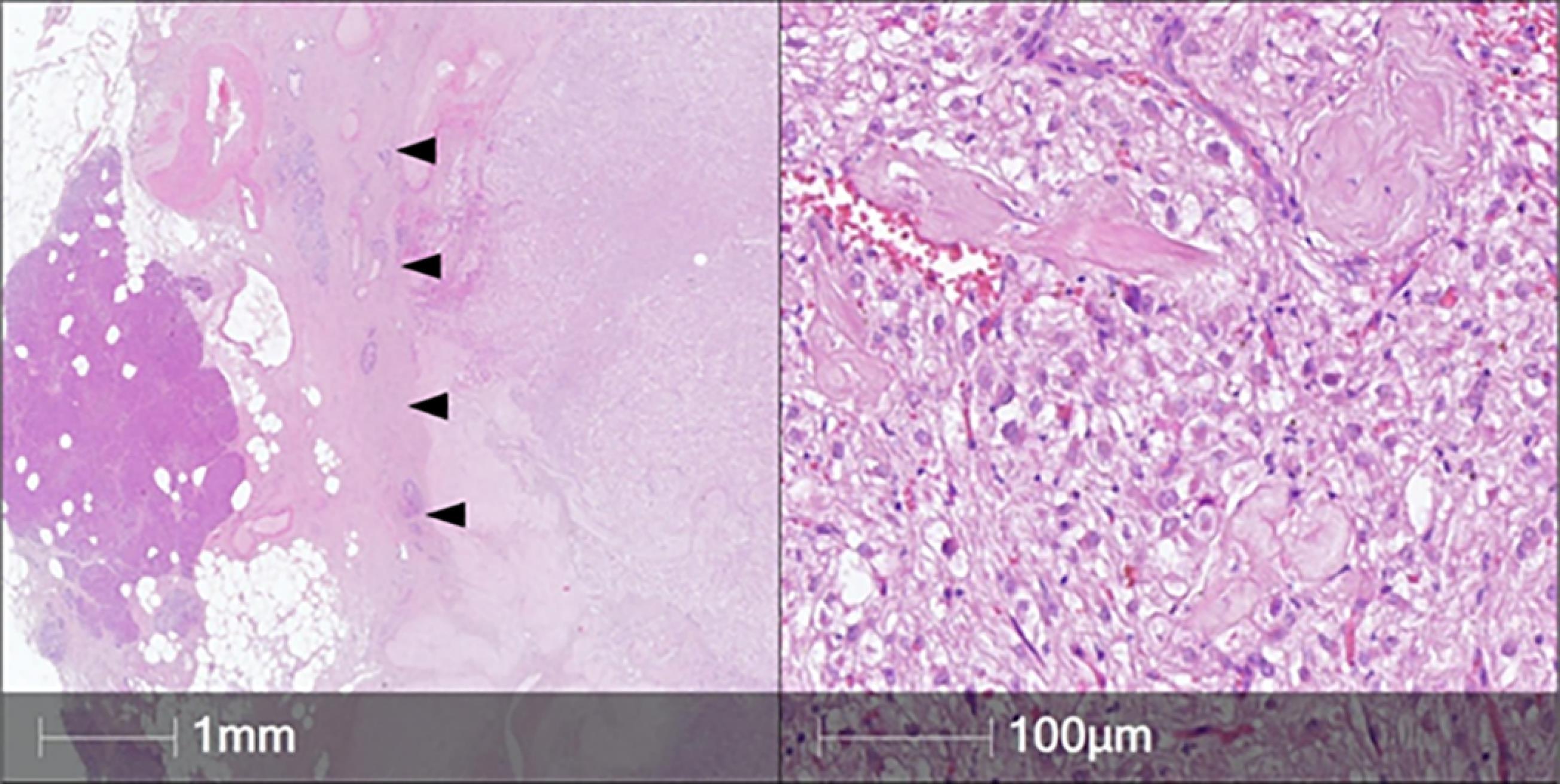
Figure 2.
Histology of the Pancreatic Tumor. Hematoxylin & eosin, original magnification × 5 (left), × 200 (right). Left: the tumor is separated from pancreatic parenchyma by a broad fibrous capsule (arrowheads). Right: large tumor cells with clear and granular cytoplasm had a relatively bland appearance
.
Histology of the Pancreatic Tumor. Hematoxylin & eosin, original magnification × 5 (left), × 200 (right). Left: the tumor is separated from pancreatic parenchyma by a broad fibrous capsule (arrowheads). Right: large tumor cells with clear and granular cytoplasm had a relatively bland appearance
Immunohistochemistry showed positivity of tumor cells for Melan A, SMA, HMB-45, TFE-3. Tumor cells were negative for SOX-10, CD34, Pan-cytokeratin, S100, CD56, Desmin (Figure 3). Proliferation index (Ki-67) measured less than 1%. These findings confirmed the diagnosis of pancreatic PEComa.

Figure 3.
Immunophenotype of the Tumor. Immunohistochemistry, chromogen – DAB, counterstaining – Mayer’s hematoxylin, original magnification × 100
.
Immunophenotype of the Tumor. Immunohistochemistry, chromogen – DAB, counterstaining – Mayer’s hematoxylin, original magnification × 100
The postoperative course was uneventful, with no signs of complications. Drainage fluid analysis showed normal amylase levels (56.16 U/L). The patient was discharged in good condition after 7 days.
Discussion
Pancreatic PEComas are exceedingly rare mesenchymal neoplasms, with only a limited number of cases reported in the literature.6
Their pathogenesis, clinical behavior, and optimal management remain incompletely understood. This case report contributes to the growing body of literature on pancreatic PEComas, emphasizing the diagnostic challenges, the role of histopathology and immunohistochemistry, and the necessity for long-term surveillance. Pancreatic PEComas often present as incidental findings or with non-specific symptoms such as vague abdominal discomfort, weight loss, or gastrointestinal disturbances. In our case, the patient experienced intermittent left upper quadrant pain over two years, an insidious presentation that aligns with prior reports.6 Given their rarity and the absence of pathognomonic radiologic features, pancreatic PEComas are frequently misdiagnosed preoperatively. Contrast-enhanced imaging typically reveals a hypervascular lesion, often leading to an initial impression of a neuroendocrine tumor or another hypervascular pancreatic neoplasm.7 Although EUS-FNA has been reported as a valuable minimally invasive method for preoperative tissue diagnosis of pancreatic masses, its role in PEComas is limited due to the rarity of the lesion and the difficulty in obtaining adequate material for definitive immunohistochemical evaluation. In several published cases, cytological smears were inconclusive or misleading, and only the cell block preparation with extended immunohistochemical panel raised suspicion of PEComa.5,8,9 In our patient, EUS-FNA was not performed preoperatively because the lesion was small, hypervascular, and surgically accessible, and because intraoperative resection would provide sufficient tissue for definitive diagnosis without the potential risks of needle tract seeding or hemorrhage.
However, in many cases, including ours, a definitive diagnosis is only established postoperatively through histopathological and immunohistochemical evaluation.6 This highlights the ongoing challenge of distinguishing PEComas from other pancreatic tumors based on imaging alone.
Histopathological examination remains the gold standard for diagnosing pancreatic PEComa. The tumor in our case exhibited the classical features described in the literature: a proliferation of epithelioid and spindle cells with abundant clear and granular cytoplasm, a rich vascular network and low mitotic activity. These findings are consistent with prior reports, reinforcing the characteristic histological profile of PEComas. Immunohistochemically, PEComas demonstrate dual melanocytic and smooth muscle differentiation. Specifically, the combination of strong HMB-45, Melan-A, and SMA positivity, together with negativity for SOX-10, CD34, pan-cytokeratin, and S100, effectively rules out most histological mimickers.10 For example, clear cell carcinoma of the pancreas or metastatic renal cell carcinoma will show cytokeratin expression, Gastrointestinal stromal tumors are typically possitive for CD117 and DOG1 positive, melanomas are S100 and SOX-10 positive, and leiomyosarcomas express SMA and desmin but lack melanocytic markers. This underlines the diagnostic value of a broad immunohistochemical panel in distinguishing PEComas from other clear cell and spindle cell pancreatic neoplasms.11
Surgical resection remains the mainstay of treatment for pancreatic PEComas, particularly in cases where the malignant potential is uncertain. In our case, a distal subtotal pancreatectomy with splenectomy was performed, consistent with standard surgical approaches for pancreatic tail tumors.12 The patient’s postoperative course was uneventful, and she was discharged in stable condition, mirroring outcomes in other reported cases where complete resection resulted in a favorable short-term prognosis. Despite their often indolent behavior, the biological potential of pancreatic PEComas remains a subject of debate. While many cases exhibit benign behavior, others demonstrate aggressive features, including local recurrence and distant metastasis.13 The risk stratification criteria proposed by Folpe et al suggest that PEComas with a size larger than 5 cm, high mitotic rate, necrosis, vascular invasion, or infiltrative growth may have a higher malignant potential.14 In our case, the tumor measured 3 cm, had low mitotic activity, and lacked necrosis or vascular invasion, suggesting a low risk of malignancy. However, given the unpredictable nature of PEComas, long-term follow-up is warranted.
Conclusion
Pancreatic PEComa is a rare and diagnostically challenging entity. Its non-specific clinical presentation and imaging characteristics require a high index of suspicion, with definitive diagnosis depend on histopathological and immunohistochemical confirmation. Surgical resection remains the primary treatment for pancreatic PEComas, offering favorable outcomes in most cases. However, due to the unpredictable biological behavior of these tumors, long-term surveillance is essential. As more cases are documented, a clearer understanding of their clinical course, molecular characteristics, and optimal management strategies will emerge. This progress will pave the way for standardized diagnostic and therapeutic guidelines.
Competing Interests
The authors declare no competing interests.
Ethical Approval
This research was conducted with the approval of the Ethics Committee of Gomel State Medical University, following all relevant ethical guidelines and regulations. Written informed consent was obtained from the patient.
Funding
This work is not supported by any funding from a company or granting agency, nor is it paid to the authors. Equipment explicitly used for the case described is also not billed to the patient, and laboratory tests are not billed to the patient. Support was provided solely from institutional and departmental source.
References
- Hirabayashi K, Nakamura N, Kajiwara H, Hori S, Kawaguchi Y, Yamashita T. Perivascular epithelioid cell tumor (PEComa) of the pancreas: immunoelectron microscopy and review of the literature. Pathol Int 2009; 59(9):650-5. doi: 10.1111/j.1440-1827.2009.02421.x [Crossref] [ Google Scholar]
- Zamboni G, Pea M, Martignoni G, Zancanaro C, Faccioli G, Gilioli E. Clear cell “sugar” tumor of the pancreas A novel member of the family of lesions characterized by the presence of perivascular epithelioid cells. Am J Surg Pathol 1996; 20(6):722-30. doi: 10.1097/00000478-199606000-00010 [Crossref] [ Google Scholar]
- Zemet R, Mazeh H, Neuman T, Freund HR, Eid A. Asymptomatic pancreatic perivascular epithelial cell tumor (PEComa) in a male patient: report and literature review. JOP 2011; 12(1):55-8. [ Google Scholar]
- Gondran H, Thebaud E, Moreau A, Le Rhun M, Touchefeu Y, Regenet N. First pancreatic perivascular epithelioid cell tumor (PEComa) treated by mTOR inhibitor. Pancreatology 2019; 19(4):566-8. doi: 10.1016/j.pan.2019.05.459 [Crossref] [ Google Scholar]
- Collins K, Buckley T, Anderson K, Karasik M, Ligato S. Perivascular epithelioid cell tumor (PEComa) of pancreas diagnosed preoperatively by endoscopic ultrasound-guided fine-needle aspiration: a case report and review of literature. Diagn Cytopathol 2017; 45(1):59-65. doi: 10.1002/dc.23599 [Crossref] [ Google Scholar]
- Geng C, Cao Z, Bacacao B, Cao Z. Perivascular epithelioid cell tumor of the pancreas: case report and literature review. Int J Clin Exp Pathol 2021; 14(5):653-61. [ Google Scholar]
- Umesh RN, Janakiraman S, Thangasamy S, Sathyanesan J. Perivascular epithelioid cell tumor of pancreas on the background of chronic liver disease: a case report and review of literature. Int Surg J 2023; 10(8):1412-8. doi: 10.18203/2349-2902.isj20232344 [Crossref] [ Google Scholar]
- Tsukita H, Koyama K, Ishinari T, Takahashi A, Miyabe K, Umakoshi M. A case of pancreatic PEComa with prominent inflammatory cell infiltration: the inflammatory subtype is a distinct histologic group of PEComa. Diagn Pathol 2024; 19(1):59. doi: 10.1186/s13000-024-01485-2 [Crossref] [ Google Scholar]
- Mohamadnejad M, Eloubeidi MA. Cystsic lesions of the pancreas. Arch Iran Med 2013; 16(4):233-9. [ Google Scholar]
- Nogueira Sixto M, Carracedo Iglesias R, Estévez Fernández S, Rodríguez Pereira C, Sánchez Santos R. Pancreatic PEComa, a not so uncommon neoplasm? Systematic review and therapeutic update. Gastroenterol Hepatol 2024; 47(1):93-100. doi: 10.1016/j.gastrohep.2023.05.009 [Crossref] [ Google Scholar]
- Thompson ED, Hruban RH, Oshima K, Epstein JI. Differential Diagnoses in Surgical Pathology: Pancreatic and Biliary Pathology. 1st ed. Philadelphia: Wolters Kluwer; 2022.
- Kinzel A, McArthur M, Gettle LM, Felker E, Patel M. PEComas: a review of imaging and clinical features. Clin Imaging 2024; 116:110332. doi: 10.1016/j.clinimag.2024.110332 [Crossref] [ Google Scholar]
- Zizzo M, Ugoletti L, Tumiati D, Castro Ruiz C, Bonacini S, Panebianco M. Primary pancreatic perivascular epithelioid cell tumor (PEComa): a surgical enigma A systematic review of the literature. Pancreatology 2018; 18(3):238-45. doi: 10.1016/j.pan.2018.02.007 [Crossref] [ Google Scholar]
- Folpe AL, Mentzel T, Lehr HA, Fisher C, Balzer BL, Weiss SW. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol 2005; 29(12):1558-75. doi: 10.1097/01.pas.0000173232.22117.37 [Crossref] [ Google Scholar]