Arch Iran Med. 28(10):592-598.
doi: 10.34172/aim.34458
Case Report
A Novel Mutation in DDR2 Associated with Warburg-Cinotti Syndrome in a Neonate
Junping Xiao Data curation, Formal analysis, Investigation, Validation, Writing – original draft, Writing – review & editing, 1, # 
Chenyu Zhuan Data curation, Formal analysis, Investigation, Validation, Writing – original draft, Writing – review & editing, 1, #
Lingkong Zeng Conceptualization, Methodology, Writing – review & editing, 1
Xuwei Tao Conceptualization, Methodology, Project administration, Supervision, Visualization, Writing – review & editing, 1, *
Author information:
1Department of Neonatology, Wuhan Women and Children Medical Care Center, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
#Contributed equally as first authors.
Abstract
Warburg-Cinotti syndrome (WCS) is a rare disorder caused by mutations in the DDR2 gene. We report the first neonatal case with a novel WCS variant, aiming to explore its clinical and genetic characteristics. Clinical data were collected and analyzed retrospectively, and whole exome sequencing (WES) was performed for the family. The patient exhibited significant respiratory distress due to choanal abnormalities, unlike previous reports. WES revealed a maternally inherited heterozygous missense mutation in DDR2 (c.431A>G, p.Asn144Ser). In-vitro experiments showed that the mutated DDR2 fails to activate the p38 MAPK pathway. The study suggests that this novel mutation may contribute to the patient’s condition, especially in the neonatal period, and may expand the phenotypic spectrum, providing new references for clinical diagnosis and gene therapy.
Keywords: Discoidin domain, Discoidin domain receptor 2, Neonatal respiratory distress, Warburg-Cinotti syndrome
Copyright and License Information
© 2025 The Author(s).
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article as: Xiao J, Zhuan C, Zeng L, Tao X. A novel mutation in DDR2 associated with warburg-cinotti syndrome in a neonate. Arch Iran Med. 2025;28(10):592-598. doi: 10.34172/aim.34458
Introduction
Warburg-Cinotti syndrome (WCS, MIM: 618175) is a rare inherited autosomal dominant disorder caused by mutations in the discoidin domain receptor 2 (DDR2) gene. The first reported case involved a male patient presenting with a range of symptoms, including corneal pannus, conductive hearing loss, hand joint flexion conjectures, phalangeal osteolytic defects, joint lesions, significant fat loss in the hands, feet, and face, oligospermia, spontaneous pneumothorax, and facial dysmorphism.1 This case was pivotal in recognizing WCS as a novel syndrome. Subsequently, another male patient with similar clinical features was reported.2 A comprehensive review analyzed six cases further expanded the phenotype spectrum of WCS, and confirmed its significant association with specific DDR2 missense- p.Leu610Pro and p.Tyr740Cys. These mutations, located in highly conserved regions of the DDR2 amino acid sequence, were validated through genetic functional assessments and have been implicated in various physiological and pathological conditions, including impaired ovulation, reduced sperm production, aberrant bone formation, abnormal angiogenesis, organ fibrosis, osteoarthritis, epithelial tissue tumors, and delayed wound healing.3
Our case represents the first reported instance of WCS presenting in the neonatal period, with genetic functional testing revealing a DDR2 mutation potentially associated with the condition. Our findings expand the known clinical phenotype of WCS and offer valuable insights for early diagnosis and gene therapy in neonates with this syndrome.
Case Report
The patient was a 2-day-old term male infant who presented with respiratory distress immediately after birth. The infant was delivered vaginally (G2P1) with Apgar scores of 9, 10, and 10 at 1, 5, and 10 minutes, respectively. His birth weight was 2.8 kg (10th percentile, -8.4 SD), head circumference was 36 cm (97th percentile, + 1.7 SD), and length was 53 cm (97th percentile, + 1.6 SD). The amniotic fluid, umbilical cord, and placenta were all normal. Despite initial intubation and mechanical ventilation in the local neonatal intensive care unit, the infant showed no signs of improvement after two days and was subsequently transferred to our ward.
Upon admission, the patient’s physical examination revealed a temperature of 36.8 °C, heart rate of 146 beats/min, and blood pressure of 62/46 mm Hg. Notable findings included a short chin, a short neck with evident webbing, and lower-set ears. The thoracic cavity appeared well-developed, but decreased breath sounds were noted in the left lung compared to the right. A grade-I continuous systolic murmur was detected in the precordial area. Muscle tone in the limbs was elevated, with restricted voluntary movements. The penis measured approximately 5 mm (-4.25 SD) in length. A soft, solid mass measuring 6 × 5 mm was palpated in the sacral region, with overlying skin showing no sinus tracts, hair coverage, discharge, or signs of erythema, swelling, heat, or pain. These abnormalities are illustrated in Figure 1.
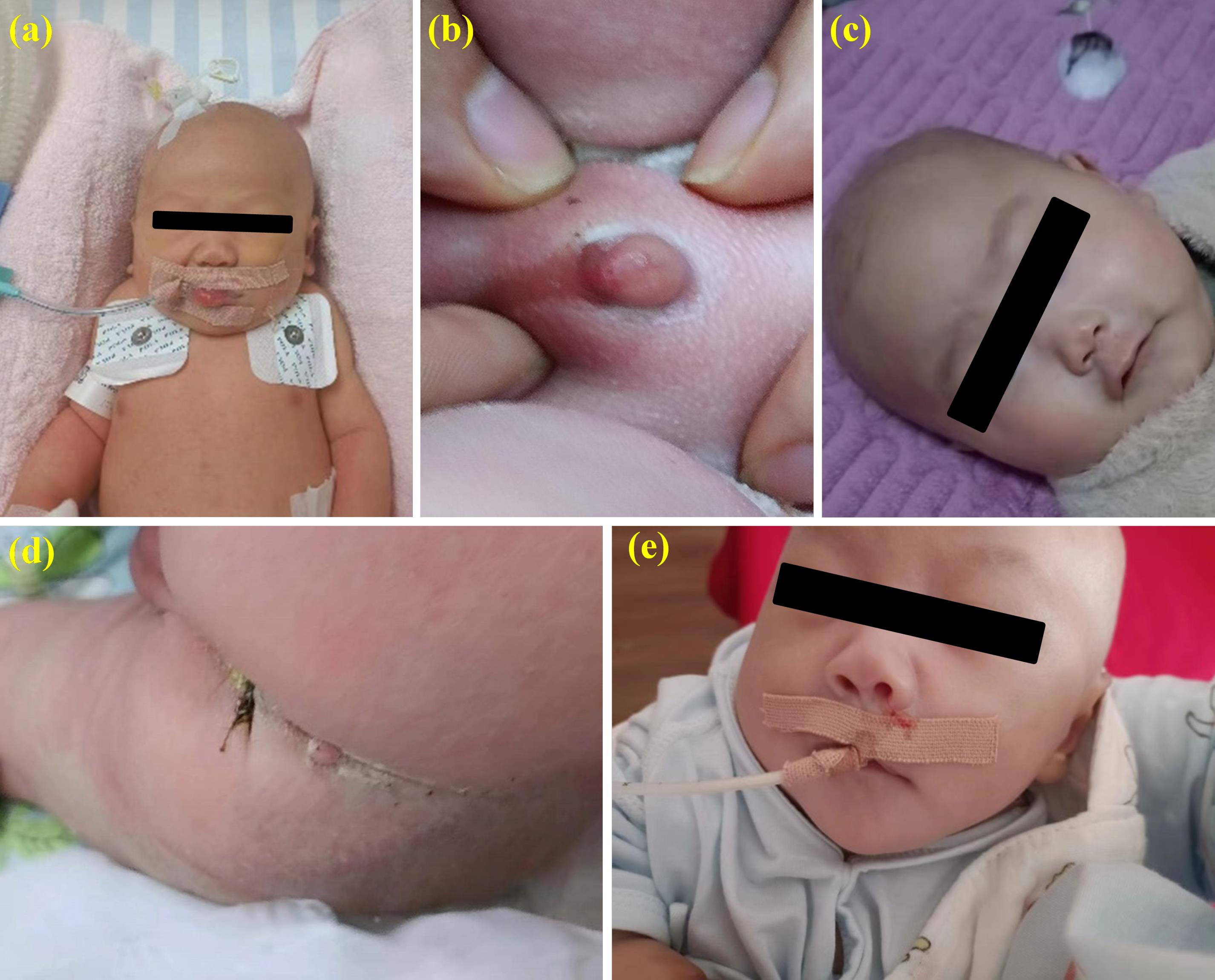
Figure 1.
Physical Characteristics of the Case. (a) Short neck with webbing; (b) Microphallus; (c) Low nasal bridge; (d) Mass at the sacral region; (e) Lower-set ears
.
Physical Characteristics of the Case. (a) Short neck with webbing; (b) Microphallus; (c) Low nasal bridge; (d) Mass at the sacral region; (e) Lower-set ears
Laboratory investigations, including complete blood count, biochemical parameters, blood amino acids, urine organic acids, and cultures of blood and sputum, revealed no significant abnormalities. Ultrasound imaging indicated the presence of an atrial septal defect measuring approximately 2.4 mm, potential hypoplastic testicles, and laxity in the left hip joint. CT scans revealed nasal malformations, specifically left choanal atresia and right posterior nasal stenosis, which were further confirmed by fiberoptic bronchoscopy and cranial MRI. Additionally, distal enlargement of the right 6th and 7th ribs was noted on CT imaging (Figure 2).
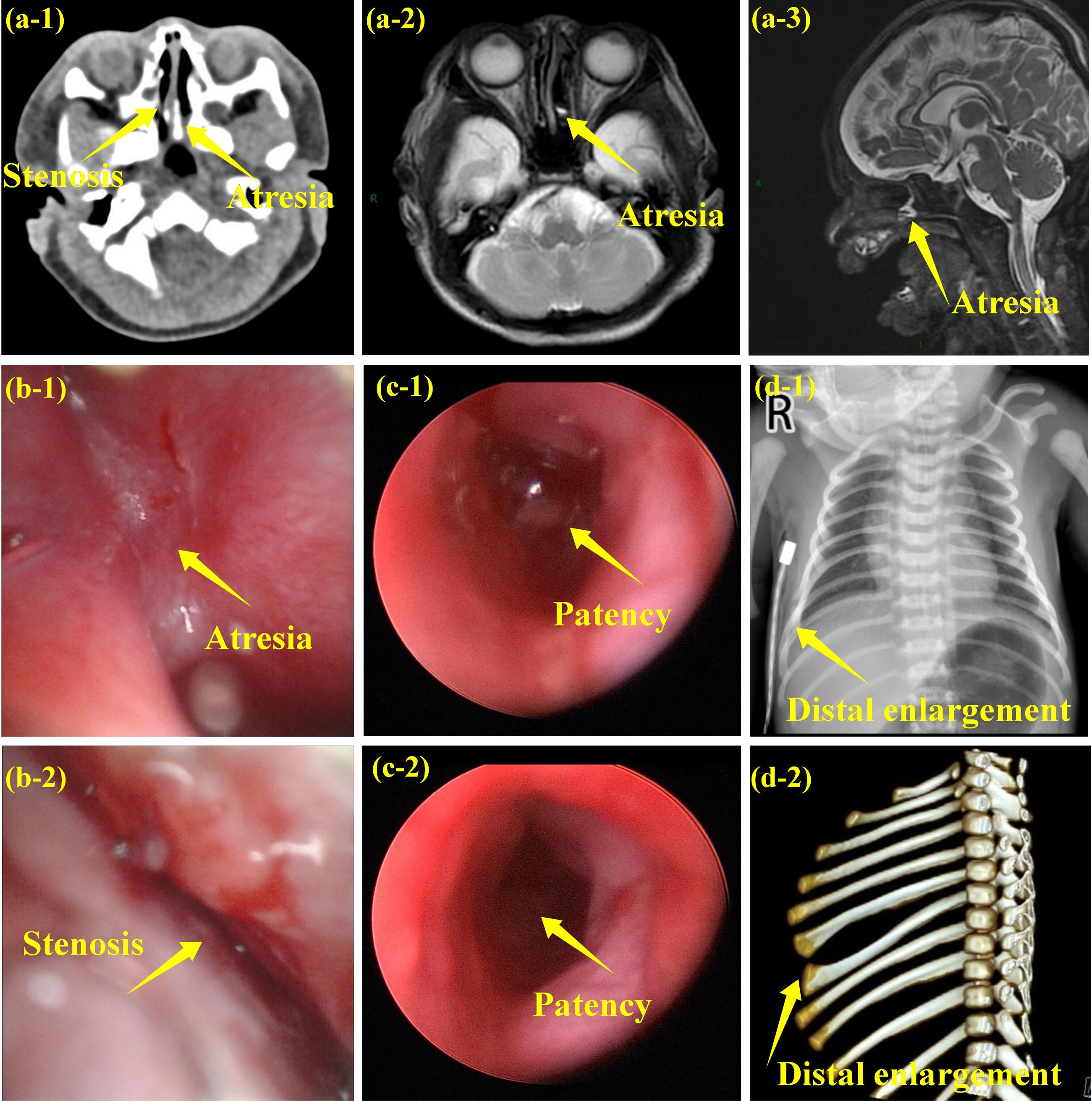
Figure 2.
Imaging Findings of the Case.(a) Nasal abnormalities by radiographic examination: left choanal atresia and right posterior nasal stenosis (a-1: CT-scan, a 2-3: MRI);(b) Nasal abnormalities by fiberoptic bronchoscopy (b1-2: after birth, b3-4: after operation);(c) Distal enlargement of the right 6th and 7th ribs (c-1: X-ray, c-2: CT-scan)
.
Imaging Findings of the Case.(a) Nasal abnormalities by radiographic examination: left choanal atresia and right posterior nasal stenosis (a-1: CT-scan, a 2-3: MRI);(b) Nasal abnormalities by fiberoptic bronchoscopy (b1-2: after birth, b3-4: after operation);(c) Distal enlargement of the right 6th and 7th ribs (c-1: X-ray, c-2: CT-scan)
Whole exome sequencing (WES) identified a maternally inherited heterozygous missense genomic variant, NM_006182.4(DDR2):c.431A > G, resulting in an amino acid substitution p.(Asn144Ser) (Figure 3). The affected asparagine residue at position 144 is located within the N-terminal of DDR2, which is highly conserved across vertebrate and invertebrate homologs. This case involves a maternally inherited DDR2 mutation. The mother has congenital polydactyly but presents with no other abnormalities. The mother exhibited mild or no symptoms (incomplete penetrance), suggesting that maternal modifiers and environmental factors may significantly mitigate the phenotypic severity of this mutation.
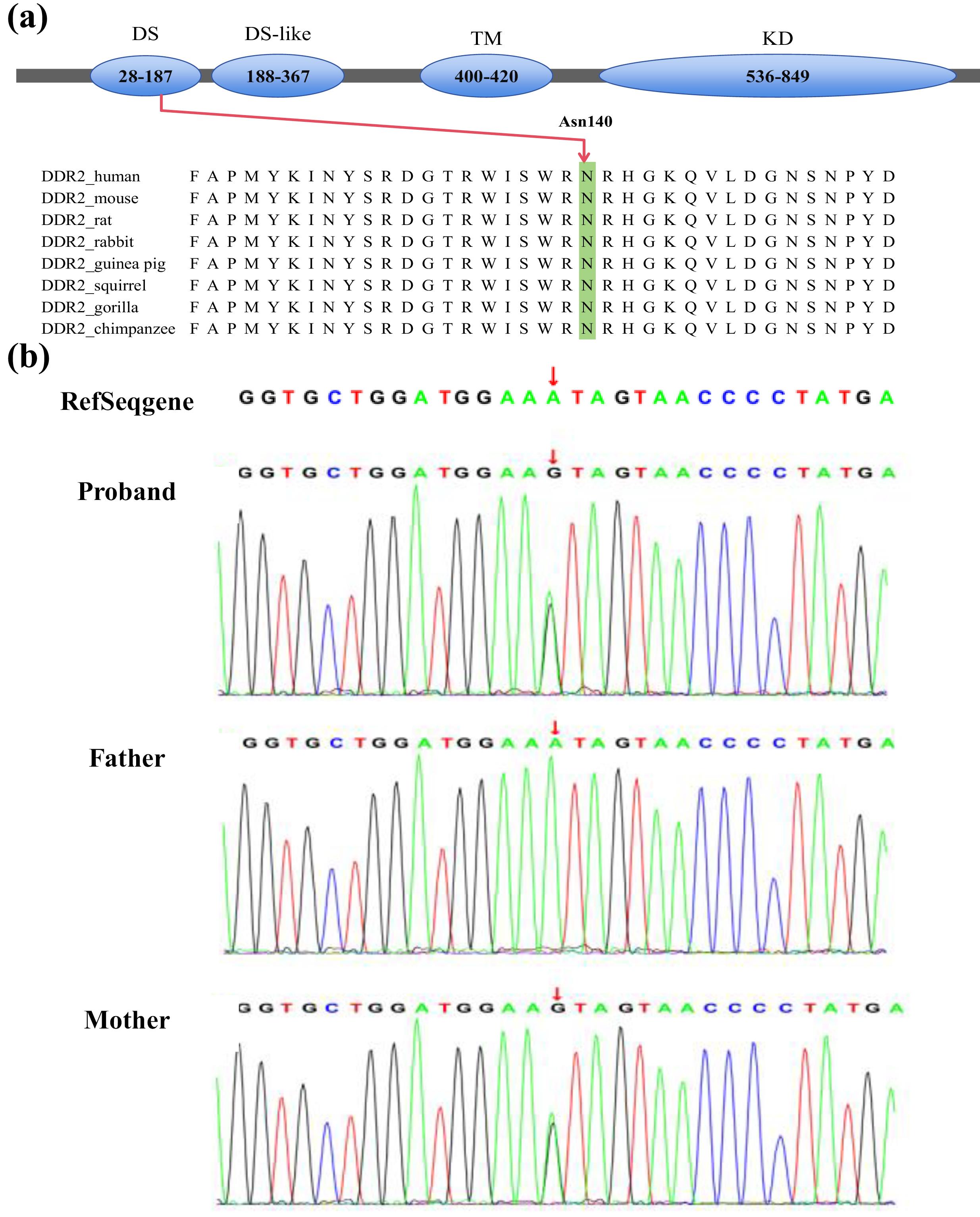
Figure 3.
(a) Domains structure diagram of DDR2; (b) Whole exome sequencing of the family
.
(a) Domains structure diagram of DDR2; (b) Whole exome sequencing of the family
Upon admission, the patient was initiated on mechanical ventilation and received comprehensive supportive care, including anti-infective therapy and enteral nutrition. (Video S1 shows the respiratory distress; see Supplementary file 1). Following the diagnosis of choanal deformities the patient underwent surgical intervention on the 10th day of hospitalization (Figure 2) and was discharged after 25 days of treatment. During the 9-month follow-up, the patient developed milia-sized hypopigmented macules on both hands, accompanied by erythema, papules, and scratch marks on both knee joints, as well as rib flaring. Developmental delays were noted, with measurements indicating a weight of 8100 g (5th-10th percentile), head circumference of 41.9 cm (less than 3rd percentile), and height of 67.5 cm (3rd percentile). Unfortunately, the parents declined to provide any additional information regarding the patient’s condition.
Previous studies have reported that DDR2 is upstream of the MAPK and PI3K/Akt pathways, suggesting that altered expression or loss of function of DDR2 impacts these pathways. Based on these findings, we conducted plasmids with wild-type DDR2 or mutant DDR2 carrying the c.431A > G mutation (Mut-DDR2) (Figure 4a), and then transfected them into HEK293WT cells for exploring the differential protein expression. Plasmids with wild type DDR2 (wt-DDR2) (NM_006182) and mutated DDR2 c.431A > G gene were synthesized by AuGCT Biotech (Beijing). HEK293wt cells were transiently transfected with the recombinant plasmids pcDNA-wt/mutated DDR2 using Liposomal Transfection Reagent (Lipofectamine 3000, Thermo Fisher Scientific) as per instructions. After transfection, the cells were cultured in fresh DMEM for 48 h, after which total protein was extracted. The proteins were blotted on a nitrocellulose membrane and then incubated with primary antibodies. The membranes were then washed and incubated with a horseradish peroxidase-conjugated anti-mouse antibody or peroxidase-conjugated anti-rabbit antibody (1:1,000; Beyotime, Shanghai, China). The proteins were detected using ECL reagents (Biosharp, Beijing, China). Densitometric evaluation was performed using Image J (NIH, United States). All data are expressed as mean ± SEM, and unpaired t-test was used to assess statistical significance between the two groups. Statistics were computed with GraphPad Prism 8 (GraphPad Software). P < 0.05 was considered as statistically significant. As shown in Figures 4b and 4c, only p38 MAPK was activated by DDR2 overexpression, but not AKT or JNK MAPK pathways. Meanwhile, Mut-DDR2 caused a decrease in p38 MAPK activation, indicating a loss of function associated with the mutation.
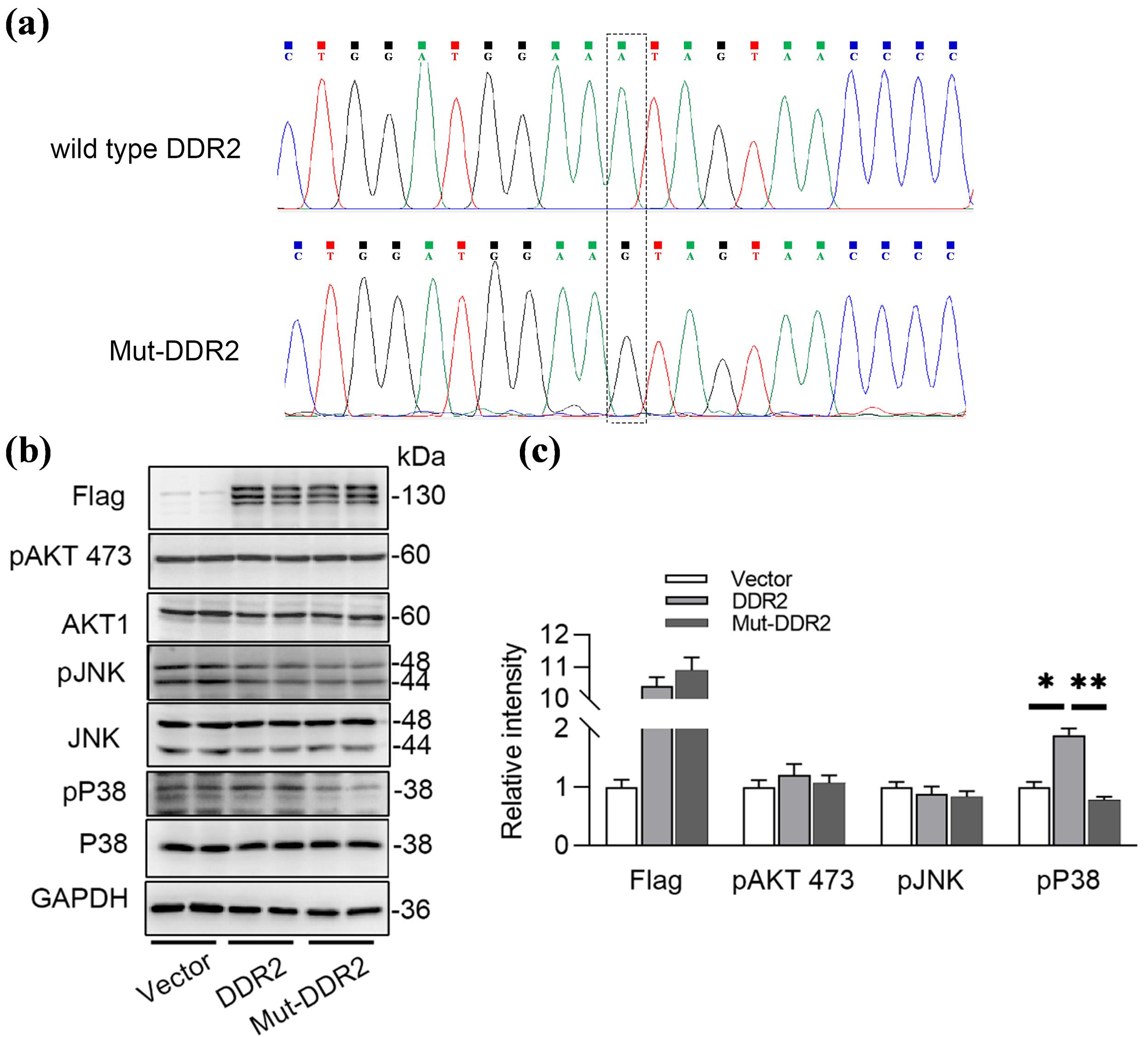
Figure 4.
Mutated DDR2 Failed to Activated the MAPK Pathway.(a) Wild type or mutated DDR2 (c.431A > G) was constructed into pcDNA3.1 vector; (b, c) Over-expressing wild type DDR2 or mutated DDR2 in HEK293wt cells for 48 hours, and examined the AKT and MAPK pathway using Western blot and its statistical graph. *P < 0.05, ** P < 0.01
.
Mutated DDR2 Failed to Activated the MAPK Pathway.(a) Wild type or mutated DDR2 (c.431A > G) was constructed into pcDNA3.1 vector; (b, c) Over-expressing wild type DDR2 or mutated DDR2 in HEK293wt cells for 48 hours, and examined the AKT and MAPK pathway using Western blot and its statistical graph. *P < 0.05, ** P < 0.01
We also conducted a literature search on PubMed using the following strategy: ([Discoidin Domain Receptor] OR [Discoidin Domain]) OR [Discoidin Domain Receptor 2] AND [Discoidin Domain Receptor 2 mutation] on July 18, 2024. This search yielded a total of 70 articles. We manually reviewed these articles to identify patients who met the inclusion criteria for WCS. Additionally, we examined the cross-references of relevant articles to ensure comprehensiveness. The final reference list was curated based on relevance to the topic under review. Only articles published in English were included in our review. We identified six previously published cases across three articles between 2006 and 2024. Including the one new case presented in this study, a total of seven cases were available for analysis. Mutations c.1829T > C (p.Leu610Pro) and c.2219A > G (p.Tyr740Cys) in DDR2 have been documented.1,2,4,5 These mutations affect amino acid residues at intracellular tyrosine binding sites, leading to increased autophosphorylation of DDR2, which is hypothesized to underlie the pathophysiology of WCS. Previous cases did not present with clinical symptoms immediately after birth; rather, the symptoms emerged gradually, encompassing skin, joint, ocular, and facial deformities. The genotype and phenotype of all documented patients and our case are summarized in Table 1. Our report describes the first case of WCS with an onset during the neonatal period, highlighting an atypical presentation of this rare syndrome.
Table 1.
Genotype and Clinical Phenotype of WCS
|
|
Reference
|
Age
|
Mutation
|
Inheritance
|
Parental affected
|
Eye abnormalities
|
Facial deformities
|
Skin
|
skeletal and joint abnormalities
|
Others
|
| Our case |
after birth |
|
c.431A > G(p.Asn144Ser) |
Maternally inherited |
No |
No |
Low nasal bridge, Low nasal bridge, short neck with webbing, low-set ears, short chin. Deformities of posterior nasal choana (left choanal atresia and right posterior stenosis) |
Thin skin |
laxity in Left hip joint |
Small penis, high muscle tension in limbs, 6 × 5 mm solid mass in sacral area, atrial septal defect |
| Case 1 |
Warburg et al1 |
57y |
c.1829T > C (p.Leu610Pro) |
Unknowna |
No |
Small eye fissure, corneal neovascularization, vision loss |
Small nostrils, long face, high palatal arch, retroverted ears, micrognathi, aural atresia |
Thin skin, keloid scar tissue |
Osteolysis Joint contracture |
Pneumothorax, Intestinal problems# |
| Case 2 |
Cinotti et. al2 |
58y |
c.2219A > G (p.Tyr740Cys) |
Unknown |
No |
Small eye fissure, corneal neovascularization, vision decreased |
Small nostrils, retroverted ears |
Keloid scar tissue, Palmar fibrous bands |
Joint contracture, Osteolysis |
Conductive hearing loss, mitral valve regurgitation |
| Case 3 |
Xu et al4 |
31y |
c.2219A > G (p.Tyr740Cys) |
De novo
|
No |
Corneal neovascularization, Vision decreased |
Small nostrils, retroverted ears, long face |
Thin skin, keloid scar tissue, follicular keratosis, palmar fibrous bands |
Joint contracture, osteolysis, thin ear cartilage, joint swelling |
_ |
| Case 4 |
Xu et al4 |
8y |
c.2219A > G (p.Tyr740Cys) |
Maternally inherited |
Yes |
Small eye fissure, corneal neovascularization |
Small nostrils, long face |
Thin skin, arm papules, palmar fibrous bands |
Joint contracture, thin ear cartilage, joint swelling |
_ |
| Case 5 |
Xu et al4 |
3y |
c.2219A > G (p.Tyr740Cys) |
Maternally inherited |
Yes |
Corneal neovascularization |
Small nostrils, long face |
Follicular keratosis |
Joint contracture, Joint swelling |
_ |
| Case 6 |
Xu et al4 |
35y |
c.1829T > C (p.Leu610P) |
Unknown |
No |
Small eye fissure |
Small nostrils, High palatal arch |
Thin skin, keloid scar tissue, follicular keratosis, palmar fibrous bands |
Joint contracture, osteolysis, joint swelling |
Pneumothorax, mitral valve regurgitation, intestinal problemsb, conductive hearing loss |
aThe parents were not examined by Genomic DNA sequencing.
bIntestinal problems include esophageal reflux, pyloric stenosis, chronic diverticulitis.
Discussion
Unlike previous cases, which typically manifested during early childhood with normal health at birth and distinctive joint contractures or unusual skin features emerging around 4 to 5 years of age, our case presented symptoms during the neonatal period. Notably, the infant experienced respiratory distress due to upper airway obstruction linked to nasal deformities, along with a short neck and thin skin, suggesting that abnormal bone development already occurred in utero. By nine months of age, there was evidence of generalized developmental delay, accompanied with skin lesions. These symptoms are thought to be associated with the mutation in the DDR2 gene.
DDR2 is a member of discoidin domain receptors (DDRs) family, which constitute a subfamily of receptor-type protein tyrosine kinases (PTKs), and comprises three structural domains: an extracellular domain, a trans-membrane domain, and an intracellular kinase domain (KD).6 DDR2 is predominantly expressed in fibroblasts and mesenchymal cells of various tissues, including the skeletal muscle, smooth muscle, skin, lungs, and kidneys. Its biological functions are largely determined by selective activation through collagen types I, II, III, and X.7 The discoidin domain (DS), a homologous sequence in the extracellular binding domain, is a unique structure comprising approximately 155 amino acid residues and functions as a collagen-binding site.8 Dimerization of two DDR2 molecules promotes collagen binding to DS, which triggers phosphorylation of tyrosine residues in the KD.9 These phosphorylated tyrosine residues serve as recruitment sites for downstream signaling molecules, thereby activating various signaling pathways, including the MAPK/ERK and PI3K/Akt pathways.10 Such signaling cascades regulate critical cellular biological functions, including proliferation, adhesion, migration, and extracellular matrix remodeling.11 Genetic mutations in the DS or other regions of DDR2 may disrupt its signaling pathways, thereby altering cellular behaviors.
Bone formation is a complex biological process involving intramembranous and endochondral ossification. Runx2, a key transcriptional regulator of osteoblasts, drives the expression of osteogenesis-related genes.12 DDR2 positively regulates osteoblast differentiation through ERK-Runx2 phosphorylation, thus playing a crucial role in osteoblast maturation and homeostasis.13 DDR2 knockout mice exhibit abnormal skeletal development and inhibited chondrocyte proliferation, characterized by dwarfism, short nose, and reduced bone density.14 DDR2 also regulates chondrocyte proliferation and matrix metabolism during chondrogenesis.15 It promotes the transformation of fibroblasts into myofibroblasts with high contractility and extracellular matrix (ECM) secretion capacity, and increases collagen fiber deposition in the ECM by activating downstream signaling pathways (such as p38/MAPK and ERK/MAPK).16,17 Meanwhile, abnormally accumulated collagen fibers activate DDR2, which induces up-regulation of matrix metalloproteinases (MMPs) and forms a positive feedback loop that accelerates articular cartilage destruction and functional loss.18 The clinical features of WCS, such as joint contractures and significant bone resorption in the distal phalanges of the fingers and toes, are thought to be associated with DDR2 dysfunctions. In our case, the disruption of collagen synthesis or degradation may be associated with defects in nasal structure development, severe limb movement restrictions, and developmental delay at nine months of age.
Previous cases involved mutations located in the KD, whereas our case features a mutation in the DS. The DS is a crucial component of DDRs, harboring collagen-binding sites responsible for collagen recognition and binding, which is a key step in DDRs activation.19 Upon collagen binding to the DS, conformational changes in the receptor are induced, subsequently activating the KD and initiating downstream signaling, thereby regulating cellular biological functions.9 In the present case, the mutation site is located in the DS domain, which is highly conserved in mammals. We hypothesize that this mutation may affect normal collagen-binding, disrupt the proper folding or stability of DDR2, and influence protein conformation or interactions, thereby affecting ligand binding or receptor dimerization. Furthermore, we observed that mutated DDR2 failed to activated p38 MAPK compared to wild type DDR2 in vitro, suggesting loss of function of DDR2 after mutation.
This case was the first neonatal WCS with a novel mutation in the DS of DDR2. The patient presented with significant respiratory distress due to early-life malformations of nasal structures and subsequently developed abnormalities in the skin, bones, and overall development. The absence of certain clinical signs, such as pigmented rashes and corneal neovascularization, may be attributed to the relatively short follow-up duration. This highlights the need for prolonged observation.
Conclusion
We report a neonatal case of WCS, thereby expanding the spectrum of clinical phenotype and genotype associated with this disease. Moreover, this case highlights the potential for gene therapy as a future treatment modality.
Supplementary Files
Supplementary file 1 contains Video S1 (Severe Respiratory Distress of Patient).
(mp4)
Competing Interests
The authors declared no conflict of interest.
Data Availability Statement
The datasets used and analysed during the current study are available from the corresponding author upon reasonable request.
Ethical Approval
This study was approved by the Human Research Ethics Committee of the Wuhan Women and Children Medical Care Center (2023R068-E0). The written consent was obtained from the parents of the neonate.
Funding
The study was supported by Hubei Provincial Natural Science Foundation (No.2023AF888), and the Funding for Scientific Research Projects from Wuhan Municipal Health Commission (No.WX23A91).
References
- Warburg M, Ullman S, Jensen H, Pedersen H, Kobayashi T, Russell B. Blepharophimosis, corneal vascularization, deafness, and acroosteolysis: a “new” syndrome?. Am J Med Genet A 2006; 140(24):2709-13. doi: 10.1002/ajmg.a.31543 [Crossref] [ Google Scholar]
- Cinotti E, Ferrero G, Paparo F, Papadia M, Faravelli F, Rongioletti F. Arthropathy, osteolysis, keloids, relapsing conjunctival pannus and gingival overgrowth: a variant of polyfibromatosis?. Am J Med Genet A 2013; 161A(6):1214-20. doi: 10.1002/ajmg.a.35908 [Crossref] [ Google Scholar]
- Leitinger B. Discoidin domain receptor functions in physiological and pathological conditions. Int Rev Cell Mol Biol 2014; 310:39-87. doi: 10.1016/b978-0-12-800180-6.00002-5 [Crossref] [ Google Scholar]
- Xu L, Jensen H, Johnston JJ, Di Maria E, Kloth K, Cristea I. Recurrent, activating variants in the receptor tyrosine kinase DDR2 cause Warburg-Cinotti syndrome. Am J Hum Genet 2018; 103(6):976-83. doi: 10.1016/j.ajhg.2018.10.013 [Crossref] [ Google Scholar]
- Ours CA, Biesecker LG, Darling TN. Progression of skin lesions in Warburg-Cinotti syndrome. JAAD Case Rep 2022; 20:47-9. doi: 10.1016/j.jdcr.2021.12.006 [Crossref] [ Google Scholar]
- Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell 1997; 1(1):13-23. doi: 10.1016/s1097-2765(00)80003-9 [Crossref] [ Google Scholar]
- Leitinger B. Molecular analysis of collagen binding by the human discoidin domain receptors, DDR1 and DDR2 Identification of collagen binding sites in DDR2. J Biol Chem 2003; 278(19):16761-9. doi: 10.1074/jbc.M301370200 [Crossref] [ Google Scholar]
- Ichikawa O, Osawa M, Nishida N, Goshima N, Nomura N, Shimada I. Structural basis of the collagen-binding mode of discoidin domain receptor 2. EMBO J 2007; 26(18):4168-76. doi: 10.1038/sj.emboj.7601833 [Crossref] [ Google Scholar]
- Mariadoss AV, Wang CZ. Exploring the cellular and molecular mechanism of discoidin domain receptors (DDR1 and DDR2) in bone formation, regeneration, and its associated disease conditions. Int J Mol Sci 2023; 24(19):14895. doi: 10.3390/ijms241914895 [Crossref] [ Google Scholar]
- Chen L, Kong X, Fang Y, Paunikar S, Wang X, Brown JAL. Recent advances in the role of discoidin domain receptor tyrosine kinase 1 and discoidin domain receptor tyrosine kinase 2 in breast and ovarian cancer. Front Cell Dev Biol 2021; 9:747314. doi: 10.3389/fcell.2021.747314 [Crossref] [ Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000; 103(2):211-25. doi: 10.1016/s0092-8674(00)00114-8 [Crossref] [ Google Scholar]
- Komori T. Whole aspect of Runx2 functions in skeletal development. Int J Mol Sci 2022; 23(10):5776. doi: 10.3390/ijms23105776 [Crossref] [ Google Scholar]
- Yang H, Sun L, Cai W, Gu J, Xu D, Deb A. DDR2, a discoidin domain receptor, is a marker of periosteal osteoblast and osteoblast progenitors. J Bone Miner Metab 2020; 38(5):670-7. doi: 10.1007/s00774-020-01108-y [Crossref] [ Google Scholar]
- Labrador JP, Azcoitia V, Tuckermann J, Lin C, Olaso E, Mañes S. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep 2001; 2(5):446-52. doi: 10.1093/embo-reports/kve094 [Crossref] [ Google Scholar]
- Zhang Y, Su J, Yu J, Bu X, Ren T, Liu X. An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. J Bone Miner Res 2011; 26(3):604-17. doi: 10.1002/jbmr.225 [Crossref] [ Google Scholar]
- Zhao H, Bian H, Bu X, Zhang S, Zhang P, Yu J. Targeting of discoidin domain receptor 2 (DDR2) prevents myofibroblast activation and neovessel formation during pulmonary fibrosis. Mol Ther 2016; 24(10):1734-44. doi: 10.1038/mt.2016.109 [Crossref] [ Google Scholar]
- Ruiz PA, Jarai G. Discoidin domain receptors regulate the migration of primary human lung fibroblasts through collagen matrices. Fibrogenesis Tissue Repair 2012; 5:3. doi: 10.1186/1755-1536-5-3 [Crossref] [ Google Scholar]
- Xu L, Peng H, Glasson S, Lee PL, Hu K, Ijiri K. Increased expression of the collagen receptor discoidin domain receptor 2 in articular cartilage as a key event in the pathogenesis of osteoarthritis. Arthritis Rheum 2007; 56(8):2663-73. doi: 10.1002/art.22761 [Crossref] [ Google Scholar]
- Borza CM, Pozzi A. Discoidin domain receptors in disease. Matrix Biol 2014; 34:185-92. doi: 10.1016/j.matbio.2013.12.002 [Crossref] [ Google Scholar]