Arch Iran Med. 28(1):63-66.
doi: 10.34172/aim.31746
Case Report
A Novel Candidate Gene MACF1 is Associated with Autosomal Dominant Non-syndromic Hearing Loss in an Iranian Family
Niloofar Bazazzadegan Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing, 1, * 
Mojgan Babanejad Software, 1
Susan Banihashemi Resources, 1
Sanaz Arzhangi Resources, 1
Kimia Kahrizi Validation, Visualization, Writing – review & editing, 1
Kevin TA Booth Validation, Visualization, Writing – review & editing, 2, 3
Hossein Najmabadi Project administration, Supervision, Validation, Visualization, Writing – review & editing, 1
Author information:
1Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran
2Department of Medical and Molecular Genetics, Indiana School of Medicine, Indianapolis, IN, USA
3Department of Otolaryngology-Head and Neck Surgery, Indiana School of Medicine, Indianapolis, IN, USA
Abstract
Cytoskeletal dynamics, the interplay of actin, microtubules, and septins, is a highly coordinated and tightly regulated process. Defects in the proteins involved can result in a wide range of cellular consequences. Hearing loss is the most common sensory defect and exhibits extraordinary genetic and phenotypic heterogeneity. Currently, there are more than 170 genes casually linked to non-syndromic hearing loss (NSHL), of which more than 60 are associated with autosomal dominant inheritance. Here, we add to this growing number by implicating MACF1 (OMIM # 608271), as a novel candidate gene for autosomal dominant non-syndromic hearing loss (ADNSHL). MACF1’s cytoskeleton integrator function and hair cell expression pattern lead one to believe that it is a necessary protein for hair cells. Many protein domains in MACF1 allow for dynamic interaction with the cytoskeleton. A large Iranian family segregating progressive ADNSHL was recruited for this study. The proband had bilateral mild-moderate sensorineural hearing loss and was negative for GJB2 mutations. After applying exome sequencing on the proband, a missense mutation c.1378C>T (p.His460Tyr) was found in MACF1 and co-segregated with the hearing loss in the extended family. We speculated that MACF1 mutations probably cause non-syndromic hearing loss inherited in an autosomal dominant manner. The potential functional impact of the identified variant will be investigated through further analysis.
Keywords: Autosomal dominant non-syndromic hearing loss, Iran, MACF1, Novel gene
Copyright and License Information
© 2025 The Author(s).
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article as: Bazazzadegan N, Babanejad M, Banihashemi S, Arzhangi S, Kahrizi K, Booth KTA, et al. A novel candidate gene MACF1 associated with autosomal dominant non-syndromic hearing loss in an Iranian family. Arch Iran Med. 2025;28(1):63-66. doi: 10.34172/aim.31746
Introduction
Congenital sensorineural hearing loss (HL) affects ~1 of every 1000 live births.1 This rises to 2.8 per 1000 in school-age children and to 3.5 per 1000 adolescents.2 In developed countries, it is estimated that ~80% of HL has a genetic etiology. After clinical evaluation, comprehensive genetic testing is the next best test to determine clinical actions and interventions, and to provide a definitive diagnosis. This allows for identification of the underlying genetic cause, facilitating tailored management strategies, genetic counseling, and prognosis determination.
Genetic HL displays a vast genetic allelic and phenotypic spectrum.3 Currently, comprehensive genetic testing for HL returns positive results from a 35%‒50% diagnostic rate, depending on several variables such as: phenotype, onset, inheritance pattern, and ethnicity. This diagnostic rate illustrates the complexity of providing a genetic diagnosis, and implicates the contributions of novel genes to genetic HL that have yet to be identified.4
Currently, there are more than 170 genes casually linked to non-syndromic hearing loss (NSHL), of which more than 60 are associated with autosomal dominant inheritance (AD) (org https://hereditaryhearingloss.org/). Here, we add to this list by implicating MACF1 as a possible novel gene for postlingual progressive ADNSHL.
Case Report
The proband presented to the genetics clinic at University of Social Welfare and Rehabilitation Sciences. The proband has an extensive family history of HL (Figure 1), accompanied by no other phenotypic manifestations. After obtaining informed consent, whole blood samples were collected from participating members and genomic DNA was extracted (Figure 1). Affected members of the family underwent clinical re-evaluation to rule out potential missed syndromic forms of HL. Pure tone audiometry was performed on affected individuals and revealed a mild sloping to severe HL. The HL is described as postlingual, and progressive. Individual III.3 showed a more severe HL in the low frequencies and may represent some progression of the low frequencies with age. Initially, the proband underwent GJB2 testing, which revealed no causal mutations. Subsequently, the proband underwent exome sequencing (ES) to determine the genetic cause of HL segregating in the family. After read mapping and quality filtering, the exome data was analyzed using a tiered approach. First, variants in genes causally linked to HL were reviewed and no plausible variant was identified, for either AD or autosomal recessive NSHL. Next, a broader search was employed. Variants were filtered based on minor allele frequency [ESP6500 (http://evs.gs.washington.edu/EVS), ExAC (https://exac.broadinstitute.org/), Iranome (http://www.iranome.ir/), and gnomAD (http://gnomad.broadinstitute.org/)], inheritance pattern and predicted variant consequence. Next, variants were prioritize based on in-silico predictions [PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), GERP + + , SIFT (http://sift-dna.org) and PhyloP) and classifications in public databases [OMIM (https://www.omim.org/) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/)] (Figure 2) (Supplementary file 1, Table S1).
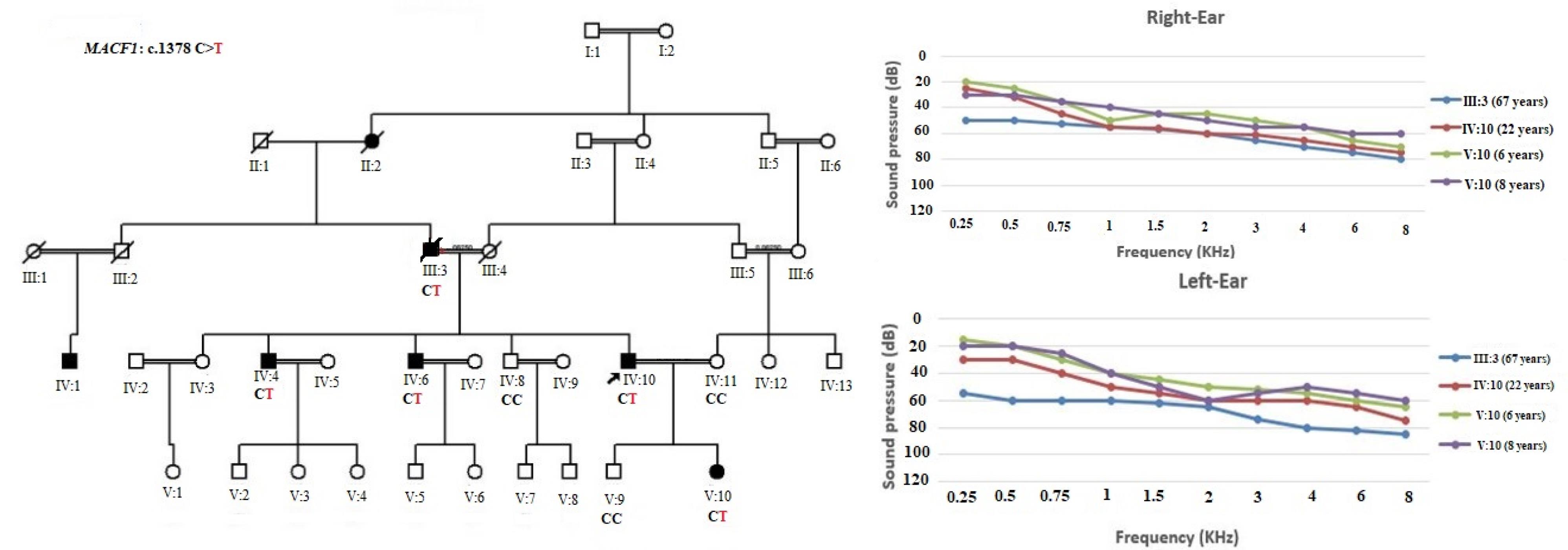
Figure 1.
Family Pedigree with Genotype of Affected and Normal Individuals (Affected individual has been shown with C/T genotype and normal individual with C/C). Pure tone audiometry for right and left ears of proband, proband’s father and daughter
.
Family Pedigree with Genotype of Affected and Normal Individuals (Affected individual has been shown with C/T genotype and normal individual with C/C). Pure tone audiometry for right and left ears of proband, proband’s father and daughter
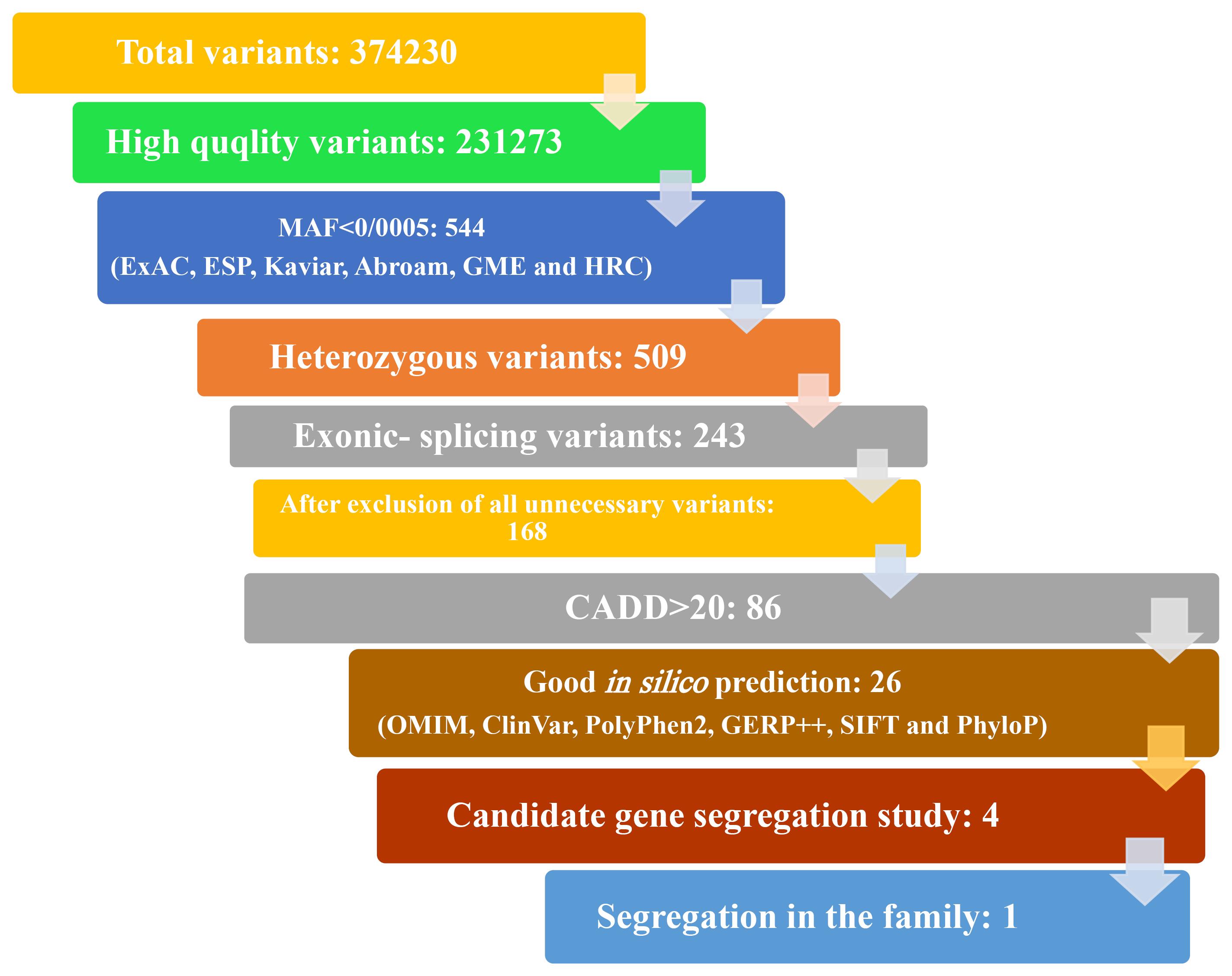
Figure 2.
Variant Filtering and Prioritization Based on In-Silico Predictions (PolyPhen2, GERP++, SIFT, and PhyloP)
.
Variant Filtering and Prioritization Based on In-Silico Predictions (PolyPhen2, GERP++, SIFT, and PhyloP)
After applying the filtering above, four variants were further prioritized for segregation within the extended family. Of these, only the heterozygote missense variant (GRCh37/hg19ch:1, 39751285, NM_012090.5, c.1378C > T; p.His460Tyr) in MACF1 co-segregated with the HL phenotype (Table 1; Figure S1). Forward primer: 5’-AGACTTCTTGGCTCCCTCTG-3’ and reverse primer: 5’-GAGTCCCTTGTTCCTCACCT-3’ were used for Sanger sequencing of the detected variant. This variant has a minor allele frequency (MAF) 0.000004 (GnomAD_exomes) and 0.000008 (ExAC). It has not been reported in Iranome which is a domestic population database.
Table 1.
Databases and In Silico Algorithms for MACF1 and Three Other Candidate Variants Undergoing Segregation Analysis
|
Gene
|
Variant
|
CADD
|
GERP
|
SIFT
|
Phylop
|
Polyphen
|
Franklin
|
InterVar
|
Clinvar
|
MIM#
|
Segregation
|
|
MACF1
|
c.1378C > T |
23.3 |
5.93 |
0.003 |
3.72 |
— |
VUS |
Likely pathogenic |
— |
608271 |
Yes |
|
DNAH14
|
c.1990-1991insTT |
— |
— |
— |
— |
— |
Likely pathogenic |
— |
— |
603341 |
No |
|
USP6
|
c.G2763A |
38 |
2.91 |
— |
9.74 |
— |
VUS |
VUS |
— |
604334 |
No |
|
MUC16
|
c.40674-40677del |
— |
— |
— |
— |
— |
VUS |
— |
— |
606154 |
No |
The histidine in position 460 was changed with tyrosine which is an aromatic amino acid. According to HOPE results (https://www3.cmbi.umcn.nl/hope/method/), the wild-type and mutant amino acids differ in size. The mutant residue is bigger which might lead to local misfolding. The hydrophobicity of the wild-type and mutant residue differs. The mutation introduces a more hydrophobic residue at this position. This can result in loss of hydrogen bonds and/or disturb correct folding.
Discussion
On human Chr 1p32, the MACF1 gene is located near the DFNA2 dominant HL locus. It has been demonstrated that HL in certain DFNA2 families is caused by mutations in KCNQ4, which are centromeric to MACF1.5-7 No plausible variants in KCNQ4 were detected in the proband based on ES analysis. The microtubule and actin crosslinking factor 1 (MACF1) gene encodes Actin Crosslinking Family Protein 7 (MACF1), a massive (~500 kDa) cytoskeletal crosslinking protein that interacts with F-actin and microtubules to shape cell morphology.6 Metazoan tissues have widespread expression of MACF1, indicating a notable degree of evolutionary conservation.8 MACF1’s cytoskeleton integrator function and hair cell expression pattern lead one to believe that it is a necessary protein for hair cells. The developmentally significant nature of microtubule and actin crosslinking factor 1 genes is established by the embryonic lethality of a null mutation in the mouse ortholog.9 Moreover, a mutation in zebrafish causes abnormalities in the oocyte and egg’s animal-vegetal polarity.10 Many protein domains in MACF1 allow for dynamic interaction with the cytoskeleton. Direct contact between calponin homology 1 and 2 domains and F-actin is facilitated towards the N terminus. The GSR (Gly-Ser-Arg)-repeat domain, which also interacts with microtubules, the EF hand domains, which bind calcium, and the GAS2-related protein domain, which binds with microtubules and helps microtubule stabilization, are located close to the C-terminus.9
Interestingly, although highly expressed, MACF1 is not required for normal hair cell development and maturation, and conditional knockout cKO mice for MACF1 have normal hearing at P30.11 We speculate two possibilities to explain the phenotype seen in humans but not the cKO mouse. First, it is possible that MACF1 is dispensable for normal hair cell development and maturation but might play a more important role throughout the life of the hair cell, and examination of its absences at P30 might not capture its true biological role. Second, we identified a missense variant which is hypothesized to act as a gain-of-function or dominant negative and the cKO mouse is not a good model to recapitulate this effect. It is noteworthy that MACF1 lies ~1.2 Mb downstream of KCNQ4, andno plausible variants were detected in the exonic regions of KNCQ4.
Currently, variants in MACF1 are causally linked to Lissencephaly 9 with complex brainstem malformation (LIS9). LIS9 is an autosomal dominant form of lissencephaly, which is a disorder that affects the development of the brain’s cortex. This disorder is characterized by the absence or thickening of the normal six-layered cortex, leading to disorganization. Clinically, LIS9 is associated with global developmental delay that is noticeable from infancy. Individuals with LIS9 also experience impaired intellectual development, often resulting in poor or absent speech. Additionally, abnormal or involuntary movements may be present. Brain imaging reveals malformation of the brainstem, as well as the presence of pachygyria and lissencephaly (MIM:618325). As it was obvious in our family, no syndrome was detected and only HL was found in the affected individuals.
Conclusion
In conclusion, we report a novel candidate gene (MACF1) for autosomal dominant non-syndromic hearing loss (ADNSHL). Functional studies are needed to understand the impact of the p.His460Tyr variant on MACF1 function and how this variant results in HL. Additionally, identifying more families segregating NSHL and variants in MACF1 will help provide a better understanding of MACF1 phenotypic spectrum. Finally, we have expended the phenotypic spectrum of MACF1 to include ADNSHL.
Supplementary Files
Supplementary file 1 contains Figure S1 and Table S1.
(pdf)
Acknowledgements
We express our gratitude to the patients’ family who took part in this research.
Competing Interests
No conflicts of interest are disclosed by the authors.
Ethical Approval
We certify that every author complied with ethical guidelines and had no conflicting interests while doing this research. Consent forms were acquired, and the study was given ethical permission (Institutional ethical approval number: IR.USWR.REC.1401.198).
Funding
This study was supported by the Genetics Research Center, University of Social Welfare and Rehabilitation Sciences Tehran, Iran.
References
- Korver AM, Smith RJ, Van Camp G, Schleiss MR, Bitner-Glindzicz MA, Lustig LR. Congenital hearing loss. Nat Rev Dis Primers 2017; 3:16094. doi: 10.1038/nrdp.2016.94 [Crossref] [ Google Scholar]
- Aldè M, Cantarella G, Zanetti D, Pignataro L, La Mantia I, Maiolino L. Autosomal dominant non-syndromic hearing loss (DFNA): a comprehensive narrative review. Biomedicines 2023; 11(6):1616. doi: 10.3390/biomedicines11061616 [Crossref] [ Google Scholar]
- Ali A, Tabouni M, Kizhakkedath P, Baydoun I, Allam M, John A. Spectrum of genetic variants in bilateral sensorineural hearing loss. Front Genet 2024; 15:1314535. doi: 10.3389/fgene.2024.1314535 [Crossref] [ Google Scholar]
- Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet 2016; 135(4):441-50. doi: 10.1007/s00439-016-1648-8 [Crossref] [ Google Scholar]
- Gong TW, Besirli CG, Lomax MI. MACF1 gene structure: a hybrid of plectin and dystrophin. Mamm Genome 2001; 12(11):852-61. doi: 10.1007/s00335-001-3037-3 [Crossref] [ Google Scholar]
- Coucke PJ, Van Hauwe P, Kelley PM, Kunst H, Schatteman I, Van Velzen D. Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum Mol Genet 1999; 8(7):1321-8. doi: 10.1093/hmg/8.7.1321 [Crossref] [ Google Scholar]
- Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat Genet 1998; 20(4):370-3. doi: 10.1038/3845 [Crossref] [ Google Scholar]
- Kubisch C, Schroeder BC, Friedrich T, Lütjohann B, El-Amraoui A, Marlin S. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 1999; 96(3):437-46. doi: 10.1016/s0092-8674(00)80556-5 [Crossref] [ Google Scholar]
- Kodama A, Karakesisoglou I, Wong E, Vaezi A, Fuchs E. ACF7: an essential integrator of microtubule dynamics. Cell 2003; 115(3):343-54. doi: 10.1016/s0092-8674(03)00813-4 [Crossref] [ Google Scholar]
- Gupta T, Marlow FL, Ferriola D, Mackiewicz K, Dapprich J, Monos D. Microtubule actin crosslinking factor 1 regulates the Balbiani body and animal-vegetal polarity of the zebrafish oocyte. PLoS Genet 2010; 6(8):e1001073. doi: 10.1371/journal.pgen.1001073 [Crossref] [ Google Scholar]
- Gilbert BL, Zhu S, Salameh A, Sun S, Alagramam KN, McDermott BM Jr. Actin crosslinking family protein 7 deficiency does not impair hearing in young mice. Front Cell Dev Biol 2021; 9:709442. doi: 10.3389/fcell.2021.709442 [Crossref] [ Google Scholar]