Arch Iran Med. 27(9):522-526.
doi: 10.34172/aim.28745
Case Report
A Village in the Southeastern Region of Iran Harboring the c.716T>A (p.Val239Asp) Mutation in SLC26A4
Farzane Zare Ashrafi Data curation, Formal analysis, Investigation, Writing – original draft, 1 
Saeed Dorgaleleh Data curation, Writing – original draft, 1
Raziye Rezvani Rezvandeh Formal analysis, Investigation, 1
Negar Kazemi Formal analysis, Investigation, 1
Nasrin Azizi Formal analysis, Investigation, 1
Masoud Edizadeh Software, 2, 3
Mohammad Hossein Azizi Validation, Writing – review & editing, 4
Kimia Kahrizi Validation, Visualization, 1
Hossein Najmabadi Conceptualization, Funding acquisition, Methodology, Resources, Validation, Visualization, Writing – review & editing, 1
Reza Najafipour Funding acquisition, Validation, Visualization, 1
Marzieh Mohseni Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Resources, Supervision, Visualization, 1, *
Author information:
1Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran
2Department of Bioinformatics, Genoks Genetic Diagnosis Center, Ankara, Turkey
3Ilyome Bioinformatics, Ankara, Turkey
4Academy of Medical Sciences of IR Iran, Tehran, Iran
Abstract
After GJB2, SLC26A4 is the second most common contributor to autosomal recessive nonsyndromic hearing loss (ARNSHL) worldwide. In this study, we used Exome Sequencing (ES) to present a village with 31 individuals affected by hereditary hearing loss (HHL) in southeastern Iran near the border of Pakistan. The village harbored the known pathogenic missense SLC26A4 (NM_000441.2):c.716T>A (p.Val239Asp) mutation, which has a founder effect attributed to Pakistan, Iran’s southeastern neighbor. Our findings, in addition to unraveling the molecular cause of non-syndromic hearing loss in these patients and further confirming the common ancestry and migration story between the people of this region and Pakistan, provide further insight into the genetic background of this region and highlight the importance of understanding the mutation spectrum of GJB2 and SLC26A4 in different regions to choose cost-effective strategies for molecular genetic testing.
Keywords: Exome sequencing, Founder mutation, Iran, Non-syndromic hearing loss, SLC26A4
Copyright and License Information
© 2024 The Author(s).
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article as: Zare Ashrafi F, Dorgaleleh S, Rezvani Rezvandeh R, Kazemi N, Azizi N, Edizadeh M, et al. A village in the southeastern region of Iran harboring the c.716T>A (p.Val239Asp) mutation in SLC26A4. Arch Iran Med. 2024;27(9):522-526. doi: 10.34172/aim.28745
Introduction
Hereditary hearing loss (HHL) is the second most prevalent clinical and genetically heterogeneous neurosensory disorder and health concern in Iran.1 HHL affects 1/166 Iranian individuals due to the high rate of consanguineous marriages in Iran.1 According to comprehensive studies of the Genetics Research Center (GRC) of the University of Social Welfare and Rehabilitation Sciences (USWR), Tehran, Iran, over two decades, the six most prevalent genes mutated in non-syndromic hearing loss (NSHL) Iranian patients were GJB2, SLC26A4, MYO15A, MYO7A, CDH23, and TMC1.1 SLC26A4 (MIM:605646) mutations are assumed to be the second most frequent cause of inherited hearing loss worldwide and in Iran after GJB2 mutations.2 The mutation spectrum of these two genes is ethnic-specific and varies among different populations.2 Until now, about 704 mutations have been recorded in SLC26A4 in the Human Gene Mutation Database (HGMD).3 SLC26A4 (Solute Carrier Family 26, Member 4) is a known Hearing Loss (HL) gene with 21 exons, located on 7q22.3 which encodes pendrin, an iodide–chloride transporter expressed in the thyroid, inner ear, and kidney.4,5 Homozygous or compound heterozygous mutations in SLC26A4 are associated with a broad phenotypic spectrum, from Pendred syndrome; PDS (MIM: 274600) to Deafness, autosomal recessive 4 (DFNB4), with enlarged vestibular aqueduct (EVA); DFNB4 (MIM: 600791).4 PDS, the most common form of syndromic hearing loss, is characterized by congenital sensorineural hearing loss, thyroid dysfunction, and severe-to-profound temporal bone abnormalities. Unlike PDS, DFNB4 with an EVA is recognized in patients without thyroid dysfunction or other systemic features.6,7 SLC26A4 mutations are common in different countries and regions, and some are ethnic-specific.8 Elucidating the distribution of SLC26A4 pathogenic variants in various regions of our country is of paramount importance for designing mutation-screening programs throughout the country for patients with HL, particularly those with goiter or enlarged vestibular aqueduct symptoms.8,9 To date, various SLC26A4 mutations with different frequencies have been observed in different regions of Iran.1,10 Of these, c.965dup (p.Asn322LysfsTer8) is a founder mutation originating from northwestern Iran, which further supports the ethnic and regional differences in the SLC26A4 mutation spectrum.1,4 Recently, we widely observed the known (c.716T > A; p.Val239Asp) mutation in southeastern Iran near the border of Pakistan, where the founder effect of this variant is assumed.11 Our finding in this report, in addition to unraveling the molecular cause of NSHL in two unrelated consanguineous Baluch families, provide further insight into the genetic background of this region, and may also be more proof of the founder effect of the p.Val239Asp mutation in Pakistan and the footprint of probable common ancestry and migration story.
Case Report
We identified two unrelated consanguineous Baluch families in southeastern Iran, one of which was large and multi-branched with 31 HL patients and the other with 2 affected members (Figure 1). Prior to sample collection, informed consent was obtained by the ethical committee of the USWR from each family member who agreed to participate in the study. A complete physical examination did not reveal any syndromic features and showed normal thyroid function and vision. The tympanometry test and otoscopic examination of each family indicated that the patients had severe to profound hearing loss. Eventually, the proband of each family, whose genetic screening was negative for GJB2 pathogenic variants, underwent Exome Sequencing (ES). Illumina NextSeq500 (Illumina, San Diego, California, USA) and Agilent SureSelectXT Human All Exon V6 (Agilent Technologies Inc., Santa Clara, CA, USA) were used for ES. Sequences were mapped to the UCSC hg38 human reference genome using the Burrows-Wheeler Aligner (BWA).12 VCF files were generated using the Genome Analysis Tool Kit (GATK)13 and annotation was performed using ANNOVAR.14 Variants were filtered based on their quality/coverage depth ( ≥ 3) and minor allele frequency (MAF < 0.5%). Variant prioritization was performed by considering the types of variants and bioinformatics predictions. All potentially causing variants detected by ES were validated by Sanger sequencing using an ABI 3500 Sequencer, and co-segregation studies were performed, including all available and informative family members. The primers used in this study for sequencing the candidate variants included the forward 5’-AGGTTTCTATCTCAGGCAAACA-3’ and reverse 5’-GCCCAGACTCAGAGAATGAA-3.’
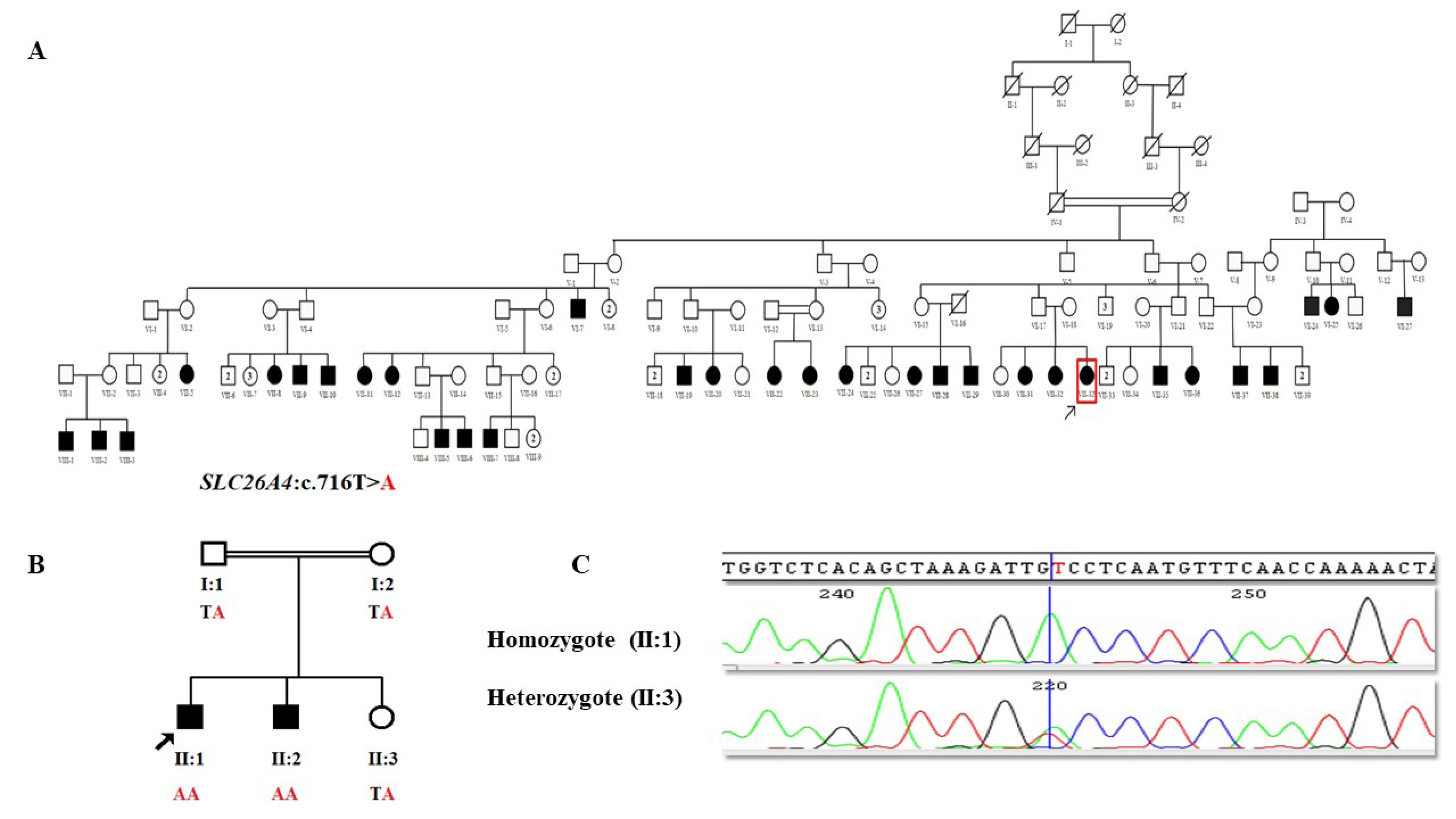
Figure 1.
A) Pedigree of family 1, B) Pedigree of family 2, C) An electropherogram of the c.716T > A mutation in one homozygote affected and one heterozygote healthy member of family 2
.
A) Pedigree of family 1, B) Pedigree of family 2, C) An electropherogram of the c.716T > A mutation in one homozygote affected and one heterozygote healthy member of family 2
By analyzing the ES data of each proband of these two families, we interestingly detected the same known pathogenic missense variant, SLC26A4 (NM_000441.2):c.716T > A (p.Val239Asp), in both families (Table 1). Sanger sequencing confirmed the presence of this pathogenic variant in their remaining affected members (Figure 1).
Table 1.
Description of the Causative Variant in These Patients
|
Gene
|
Transcript
|
Variant
|
Exon
|
SIFT
|
REVEL
|
CADD PHRED Score
|
ACMG Classification
|
|
SLC26A4
|
NM_000441.2 |
chr7:107675060:T > A
c.716T > A
(p.Val239Asp) |
6 |
Pathogenic
supporting
(0.001) |
Pathogenic
moderate
(0.935) |
27.0 |
Pathogenic |
Discussion
The total prevalence of GJB2-dependent HL in Iran is 16.5%; however, it varies in different regions of Iran from 38.3% to zero in the North and South, respectively.1 Regardless of GJB2, SLC26A4 mutations, with a frequency of 16.25%, are the highest contributors to NSHL in the Iranian population.1
In this study, we investigated the genetic cause of NSHL in two unrelated consanguineous Baluch families from the Sistan and Baluchestan province and identified SLC26A4 (NM_000441.2): c.716T > A (p.Val239Asp) mutation as a cause of HL in these families. This transversion mutation in exon 6 of SLC26A4 leads to the substitution of Valine into Aspartic Acid at position 239 of the pendrin protein which comprises 780 amino acids and contains 12 transmembrane domains. As this residue is located in the core of the transmembrane domain, the difference between the wild-type and mutant residues can disrupt the structure of this domain (Figure 2).15,16
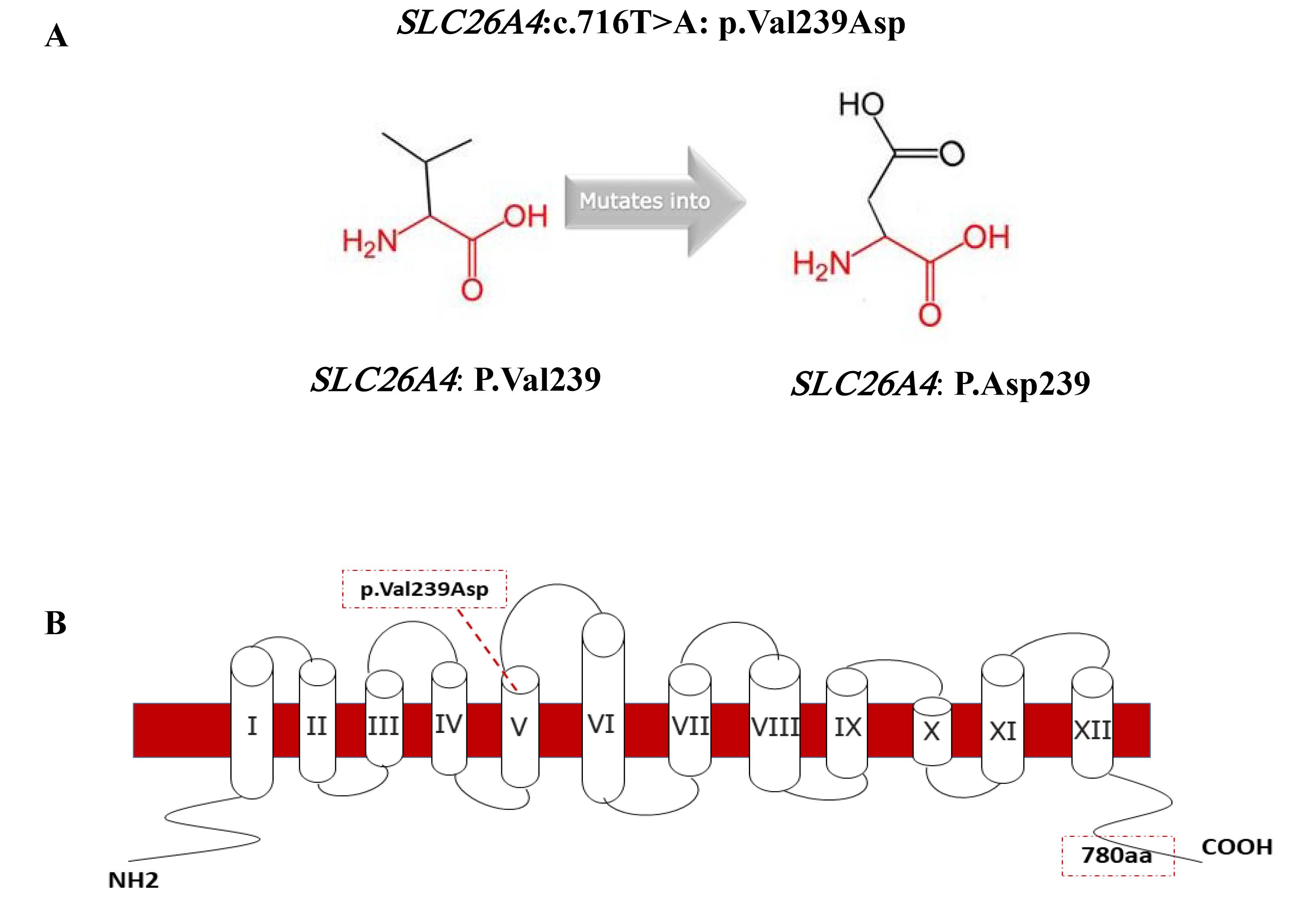
Figure 2.
A) Schematic Structures of the Original (left) and the Mutant (right) Amino Acid for the Substitution of Valine Into Aspartic (https://www3.cmbi.umcn.nl/hope/), B) The p.Val239Asp Mutation in the Schematic Model of the Human Pendrin Protein
.
A) Schematic Structures of the Original (left) and the Mutant (right) Amino Acid for the Substitution of Valine Into Aspartic (https://www3.cmbi.umcn.nl/hope/), B) The p.Val239Asp Mutation in the Schematic Model of the Human Pendrin Protein
The results of this study, especially a large multi-branch family with 31 affected members from a small village in the border areas of Iran, prompted us to conduct further research on the origin and distribution of SLC26A4 and p.Val239Asp. So far, various mutations of SLC26A4 have been reported in Iran, which include a wide range of phenotypes from “Pendred syndrome” to non-syndromic “Deafness, autosomal recessive 4, with enlarged vestibular aqueduct”. Figure 3 shows various SLC26A4 mutations responsible for autosomal recessive non-syndromic hearing loss (ARNSHL) in Iranian families until 2022.1 A detailed investigation of our cohort data revealed that in addition to the two families mentioned in this paper, we previously identified five other families harboring this mutation. Among the 48 affected members of the seven families investigated in our cohort, 38 were Baluch (Figure 4, Table 2).1,17 In a review of studies on this mutation by other groups in Iran, only one family was reported with three affected members from the Kurdistan province with Kurdish ethnicity.18
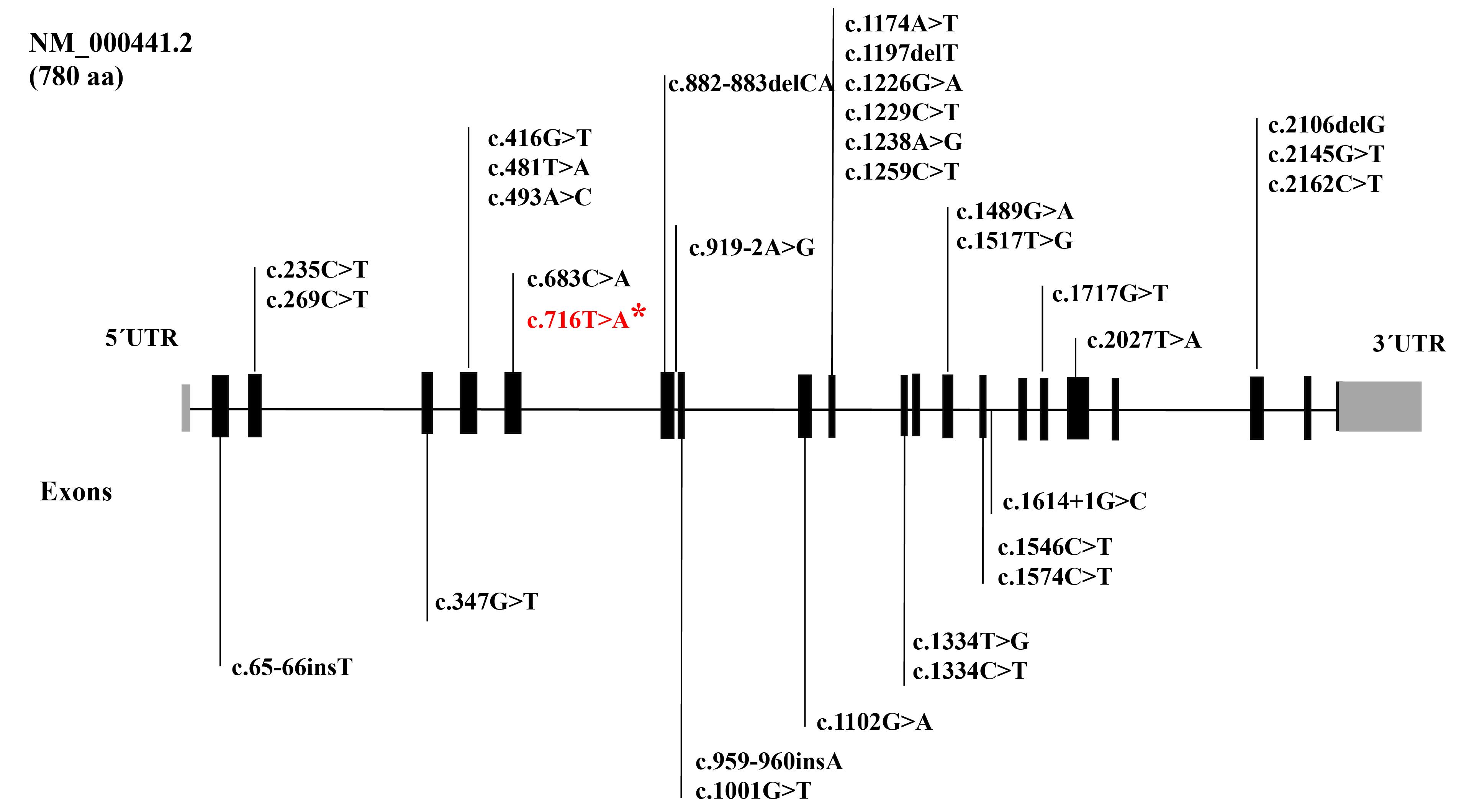
Figure 3.
SLC26A4 Mutations Responsible for ARNSHL in Iranian Families Until 2022. * The mutation identified in patients in this study
.
SLC26A4 Mutations Responsible for ARNSHL in Iranian Families Until 2022. * The mutation identified in patients in this study
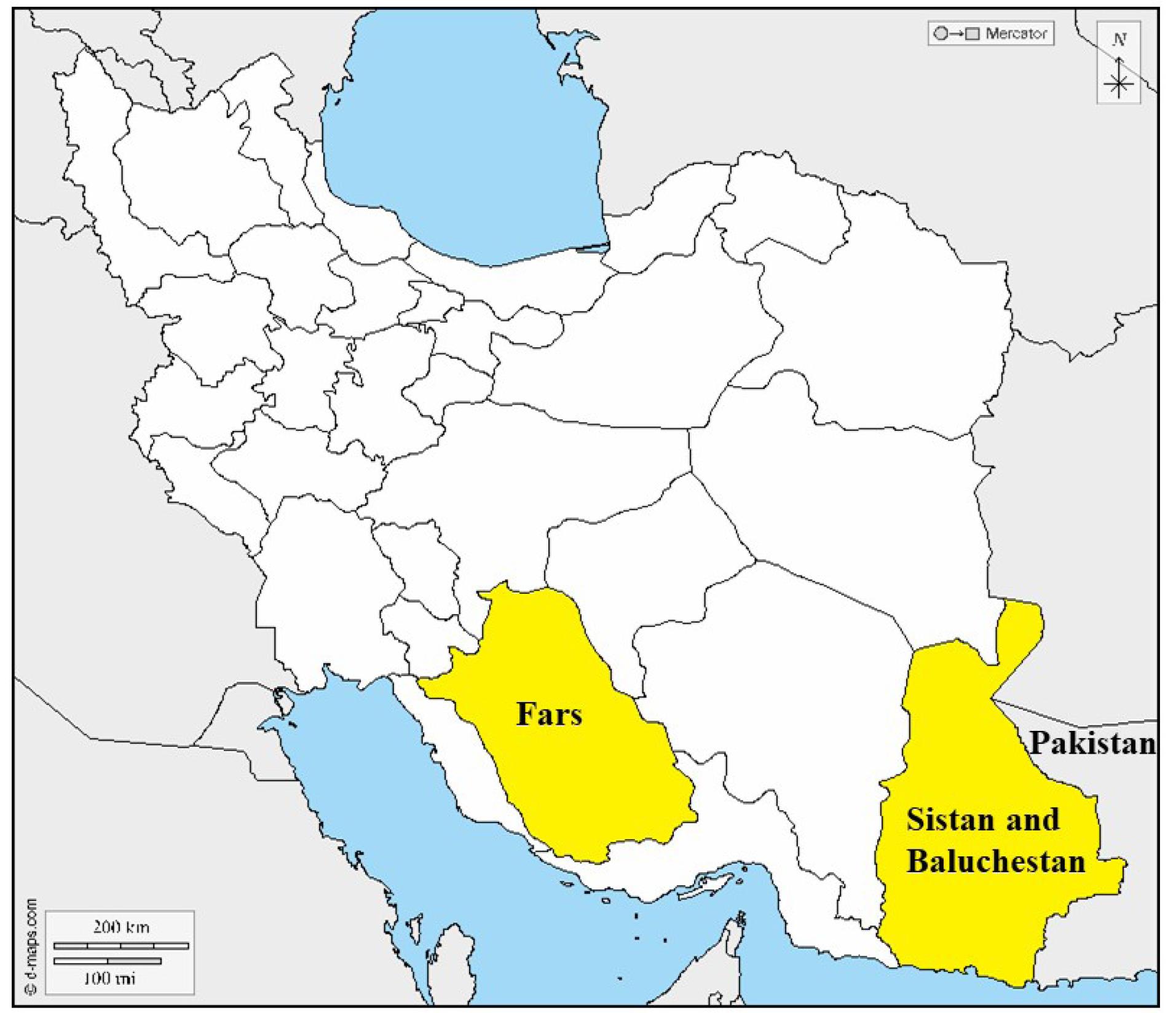
Figure 4.
Geographic Distribution of (c.716T > A; p.Val239Asp) in Our HL Cohort
.
Geographic Distribution of (c.716T > A; p.Val239Asp) in Our HL Cohort
Table 2.
Ethnicity and Number of Affected Harboring (c.716T > A; p.Val239Asp) in our HLCohort
1
|
#No
|
Family
|
Number of Affected in Pedigree
|
Variant
|
Ethnicity
|
| 1 |
Family 1 |
31 |
SLC26A4(NM_000441.2):c.716T > A (p.Val239Asp) |
Baluchis |
| 2 |
Family 2 |
2 |
Baluchis |
| 3 |
Family 3 |
4 |
Baluchis |
| 4 |
Family 4 |
1 |
Baluchis |
| 5 |
Family 5 |
3 |
Persians |
| 6 |
Family 6 |
5 |
Persians |
| 7 |
Family 7 |
2 |
Persians |
In 2015, Tsukada et al reviewed the origin and spectrum of SLC26A4 mutations worldwide and showed that p.Val239Asp is the most common mutation in Pakistan (35.6%) and Turkey (33.3%).2 Several studies have proposed a founder effect of p.Val239Asp in Pakistan.11,19,20 Finding the p.Val239Asp mutation in a large number of patients in the Sistan and Baluchestan province, which lies on the border with Pakistan, might reflect the magnification of the founder effect and contribute to the final genetic makeup of this region’s population.
Conclusion
The results of this study suggest that the gene flow of the p.Val239Asp mutation from Pakistan, a country neighboring southeastern Iran, likely changed the frequency of this mutation in that region. Given the high prevalence and mutation-specific origin of SLC26A4 mutations, considering the ethnic and regional backgrounds of patients is helpful in genetic counseling and clinical decision-making, and evaluating SLC26A4 mutations can be a first-tier screening in regions with high prevalence.
Acknowledgements
We would like to thank the patients and their families for participating in this study. Thanks also to Ms. Fatemeh Keshavarzi, Ms. Sanaz Arzhangi, Ms. Fatemeh Ghodratpour, and Mrs. Khadijeh Jalalvand from Genetics Research Centre, for their cooperation in this research.
Competing Interests
The authors declare no conflict of interest.
Ethical Approval
This study obtained ethical approval (Institutional ethical approval number: IR.USWR.REC.1401.095) and consent forms were obtained.
Funding
We acknowledge the University of Social Welfare and Rehabilitation Sciences for funding the research (Grant numbers: 2022/4/T/2821 and 2022/4/T/2834).
References
- Babanejad M, Beheshtian M, Jamshidi F, Mohseni M, Booth KT, Kahrizi K. Genetic etiology of hearing loss in Iran. Hum Genet 2022; 141(3-4):623-31. doi: 10.1007/s00439-021-02421-w [Crossref] [ Google Scholar]
- Tsukada K, Nishio SY, Hattori M, Usami S. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: their origin and a literature review. Ann Otol Rhinol Laryngol 2015; 124 Suppl 1:61s-76s. doi: 10.1177/0003489415575060 [Crossref] [ Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 2003; 21(6):577-81. doi: 10.1002/humu.10212 [Crossref] [ Google Scholar]
- Mohseni M, Honarpour A, Mozafari R, Davarnia B, Najmabadi H, Kahrizi K. Identification of a founder mutation for Pendred syndrome in families from northwest Iran. Int J Pediatr Otorhinolaryngol 2014; 78(11):1828-32. doi: 10.1016/j.ijporl.2014.08.035 [Crossref] [ Google Scholar]
- Khan MR, Bashir R, Naz S. SLC26A4 mutations in patients with moderate to severe hearing loss. Biochem Genet 2013; 51(7-8):514-23. doi: 10.1007/s10528-013-9582-0 [Crossref] [ Google Scholar]
- Kahrizi K, Mohseni M, Nishimura C, Bazazzadegan N, Fischer SM, Dehghani A. Identification of SLC26A4 gene mutations in Iranian families with hereditary hearing impairment. Eur J Pediatr 2009; 168(6):651-3. doi: 10.1007/s00431-008-0809-8 [Crossref] [ Google Scholar]
- Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 1997; 17(4):411-22. doi: 10.1038/ng1297-411 [Crossref] [ Google Scholar]
- Danilchenko VY, Zytsar MV, Maslova EA, Bady-Khoo MS, Barashkov NA, Morozov IV. Different rates of the SLC26A4-related hearing loss in two indigenous peoples of southern Siberia (Russia). Diagnostics (Basel) 2021; 11(12):2378. doi: 10.3390/diagnostics11122378 [Crossref] [ Google Scholar]
- Beheshtian M, Babanejad M, Azaiez H, Bazazzadegan N, Kolbe D, Sloan-Heggen C. Heterogeneity of hereditary hearing loss in Iran: a comprehensive review. Arch Iran Med 2016; 19(10):720-8. [ Google Scholar]
- Ghasemnejad T, Shekari Khaniani M, Zarei F, Farbodnia M, Mansoori Derakhshan S. An update of common autosomal recessive non-syndromic hearing loss genes in Iranian population. Int J Pediatr Otorhinolaryngol 2017; 97:113-26. doi: 10.1016/j.ijporl.2017.04.007 [Crossref] [ Google Scholar]
- Anwar S, Riazuddin S, Ahmed ZM, Tasneem S, Ateeq ul J, Khan SY. SLC26A4 mutation spectrum associated with DFNB4 deafness and Pendred’s syndrome in Pakistanis. J Hum Genet 2009; 54(5):266-70. doi: 10.1038/jhg.2009.21 [Crossref] [ Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010; 26(5):589-95. doi: 10.1093/bioinformatics/btp698 [Crossref] [ Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43(1110):11.0.1-.0.33. 10.1002/0471250953.bi1110s43.
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38(16):e164. doi: 10.1093/nar/gkq603 [Crossref] [ Google Scholar]
- Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 2010; 11:548. doi: 10.1186/1471-2105-11-548 [Crossref] [ Google Scholar]
- Mukherjee S, Guha M, Adhikary B, Bankura B, Mitra P, Chowdhury S. Genetic alterations in pendrin (SLC26A4) gene in adult hypothyroid patients. Horm Metab Res 2017; 49(9):680-6. doi: 10.1055/s-0043-110769 [Crossref] [ Google Scholar]
- Iran Free Map. Available from: https://d-maps.com/carte.php?num_car=5494&lang=en.
- Azadegan-Dehkordi F, Ahmadi R, Bahrami T, Yazdanpanahi N, Farrokhi E, Tabatabaiefar MA. A novel variant of SLC26A4 and first report of the c716T > A variant in Iranian pedigrees with non-syndromic sensorineural hearing loss. Am J Otolaryngol 2018; 39(6):719-25. doi: 10.1016/j.amjoto.2018.07.022 [Crossref] [ Google Scholar]
- Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet 2003; 40(4):242-8. doi: 10.1136/jmg.40.4.242 [Crossref] [ Google Scholar]
- Naz S. Molecular genetic landscape of hereditary hearing loss in Pakistan. Hum Genet 2022; 141(3-4):633-48. doi: 10.1007/s00439-021-02320-0 [Crossref] [ Google Scholar]