Arch Iran Med. 26(4):186-197.
doi: 10.34172/aim.2023.29
Original Article
Emerging Epidemiological Data on Rare Intellectual Disability Syndromes from Analyzing the Data of a Large Iranian Cohort
Farzane Zare Ashrafi Data curation, Formal analysis, Investigation, Methodology, Resources, Writing – original draft, 1, # 
Tara Akhtarkhavari Data curation, Formal analysis, Investigation, Methodology, Resources, Writing – original draft, 1, #
Zohreh Fattahi Writing – review & editing, 1
Maryam Asadnezhad Investigation, 1
Maryam Beheshtian Formal analysis, 1
Sanaz Arzhangi Resources, 1
Hossein Najmabadi Conceptualization, Visualization, Writing – review & editing, 1
Kimia Kahrizi Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Visualization, Writing – review & editing, 1, *
Author information:
1Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran
#These authors contributed equally to the work as first authors.
Abstract
Background:
Intellectual disability (ID) is a genetically heterogeneous condition, and so far, 1679 human genes have been identified for this phenotype. Countries with a high rate of parental consanguinity, such as Iran, provide an excellent opportunity to identify the remaining novel ID genes, especially those with an autosomal recessive (AR) mode of inheritance. This study aimed to investigate the most prevalent ID genes identified via next-generation sequencing (NGS) in a large ID cohort at the Genetics Research Center (GRC) of the University of Social Welfare and Rehabilitation Sciences.
Methods:
First, we surveyed the epidemiological data of 619 of 1295 families in our ID cohort, who referred to the Genetics Research Center from all over the country between 2004 and 2021 for genetic investigation via the NGS pipeline. We then compared our data with those of several prominent studies conducted in consanguineous countries. Data analysis, including cohort data extraction, categorization, and comparison, was performed using the R program version 4.1.2.
Results:
We categorized the most common ID genes that were mutated in more than two families into 17 categories. The most common syndromic ID in our cohort was AP4 deficiency syndrome, and the most common non-syndromic autosomal recessive intellectual disability (ARID) gene was ASPM. We identified two unrelated families for the 36 ID genes. We found 14 genes in common between our cohort and the Arab and Pakistani groups, of which three genes (AP4M1, AP4S1, and ADGRG1) were repeated more than once.
Conclusion:
To date, there has been no comprehensive targeted NGS platform for the detection of ID genes in our country. Due to the large sample size of our study, our data may provide the initial step toward designing an indigenously targeted NGS platform for the diagnosis of ID, especially common ARID in our population.
Keywords: Consanguinity, Epidemiology, Intellectual disability, Iran, Rare diseases
Copyright and License Information
© 2023 The Author(s).
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article as: Zare Ashrafi F, Akhtarkhavari T, Fattahi Z, Asadnezhad M, Beheshtian M, Arzhangi S, et al. Emerging epidemiological data on rare intellectual disability syndromes from analyzing the data of a large iranian cohort. Arch Iran Med. 2023;26(4):186-197. doi: 10.34172/aim.2023.29
Introduction
Intellectual disability (ID) is a frequent neurodevelopmental disorder diagnosed with cognitive and adaptive deficits before the age of 18 years.1 ID is estimated to affect 1%–3% of the global population. It can manifest as an isolated clinical manifestation or as a syndromic phenotype, as well as other physical and mental abnormalities such as behavioral problems. Based on etiology, ID can happen due to both genetic factors and pre-and post-natal environmental factors.2 Genetic factors contribute to a significant number of ID cases, and studies show that the most severe and profound ID patients are affected by monogenic disorders.2,3 Based on SysNDD (a database that contains a catalogue of published genes implicated in neurodevelopmental disorders; last update: 6/25/2022), out of 1679 genes involved in ID, 982 show an autosomal recessive (AR) mode of inheritance, 527 exhibit autosomal dominant (AD) inheritance, 154 genes show X-linked inheritance, and others are involved in ID through mitochondrial inheritance and somatic mutations.4 Prior to the advent of next-generation sequencing (NGS), the diagnosis of monogenic ID was not sufficiently fast and efficient. However, with the introduction of this technology, the identification of disease-causing variants in monogenic cases of ID has improved drastically.5 Moreover, epidemiological studies of ID in inbred countries can provide reliable data about the most prevalent ID genes or gene groups. As shown in SysNDD, autosomal recessive intellectual disability (ARID) is one of the important forms of monogenic IDs. This form of ID is a clinically and genetically extremely heterogeneous condition and has major contribution to the etiology of ID.6 It is estimated that in outbred countries, ARID accounts for about 10% of all diagnosed ID cases and contributes to 15–20% of all undiagnosed patients.6,7 At the same time, in countries with a high rate of parental consanguinity, the incidence of ARID shows a three-to four-fold increase, and rare ARIDs are more common among these populations.1,6 Although a large number of ARID genes have been identified, the abundance of these genes remains unrecognized, and there is no extensive targeted NGS platform for diagnosing ARIDs with a high confidence rate.6 Countries with a high rate of parental consanguinity provide an excellent opportunity for identification of the remaining novel genes involved in ARIDs. Since Iran is a Middle Eastern country with a parental consanguinity rate of approximately 40%, it provides a suitable population reservoir for the epidemiological study of IDs, especially ARIDs.1 The main goal of this study was to investigate the prevalence of genes identified using NGS in a large ID cohort at the Genetics Research Center of the University of Social Welfare and Rehabilitation Sciences. To the best of our knowledge, there is no comprehensive targeted NGS platform to detect ID genes in our country; therefore, considering the large sample size of this cohort, the present study may be the first step towards the design of an NGS platform for the diagnosis of ID in our country. We also compared the results of our study with those of several similar studies from other groups in consanguineous families originating from the Middle East to investigate overlapping gene defects with neighboring countries.
Materials and Methods
The epidemiological data obtained for this study were extracted from unpublished data and articles previously published by our research team.1,8-10 In order to develop the cohort, we established a genetic counseling network from all 31 provinces of Iran to include all ethnic groups in our country. Iranian families were referred by physicians or clinical geneticists from all over the country.11 The above-mentioned cohort consisted of a total of 1295 Iranian families who were referred to the Genetics Research Center of the University of Social Welfare and Rehabilitation Sciences (Iran) between 2004 and 2021 to identify genetic causes of ID. We performed total population sampling on our Iranian ID cohort. We defined the exclusion criteria as follows: families with chromosomal abnormalities, families with Fragile X syndrome, and inconsistent families. In 2011, our team studied 136 consanguineous families and applied homozygosity mapping, exon enrichment and targeted next generation sequencing.9 In another study, we performed whole-genome sequencing and/or whole exome-sequencing on 404 consanguineous families;1 it should be mentioned that these families also included undiagnosed families from our previous study. In 2019, we applied whole exome-sequencing to 100 sporadic ID cases.8 We also added ID families from the unpublished data. In total, we had 619 Iranian families with ID with definitive diagnoses of the genetic causes of this disorder. To identify the most prevalent genes in our cohort, data extraction was performed using the R program version 4.1.2.
We also compared the most prevalent genes with multiple papers that published their ID cohorts. Since Iran has a high consanguinity rate, we chose papers from countries with high rates of consanguinity. These include papers from Pakistan and the Arabs of West Asia and North Africa.12-18 Table S1 lists the genes used for the comparison. In the comparison of genes among the three groups, the following items were excluded.
-
Families with copy number variations
-
Families with multiple candidate genes
-
Samples that were investigated by a method other than NGS
We should mention that in this study, we did not have any information about ethnicity groups in other ID cohort papers, so we could not compare our data of ethnicity groups with the same ethnicity in neighboring countries.
Results
Out of 619 of the 1295 families in our ID cohort, we found 56 families that were reported twice in our cohort (56 families with mutations in 36 genes) and 65 families with a gene that was reported at least three times within the cohort (65 families with mutations in 17 genes). Based on the function of the genes, we categorized our most common genes, as depicted in Figure 1, and the number of families with mutations in each category is shown in Figure 2.
Figure 1.
The Most Commonly Reported Genes or Gene Groups in our Cohort.
.
The Most Commonly Reported Genes or Gene Groups in our Cohort.
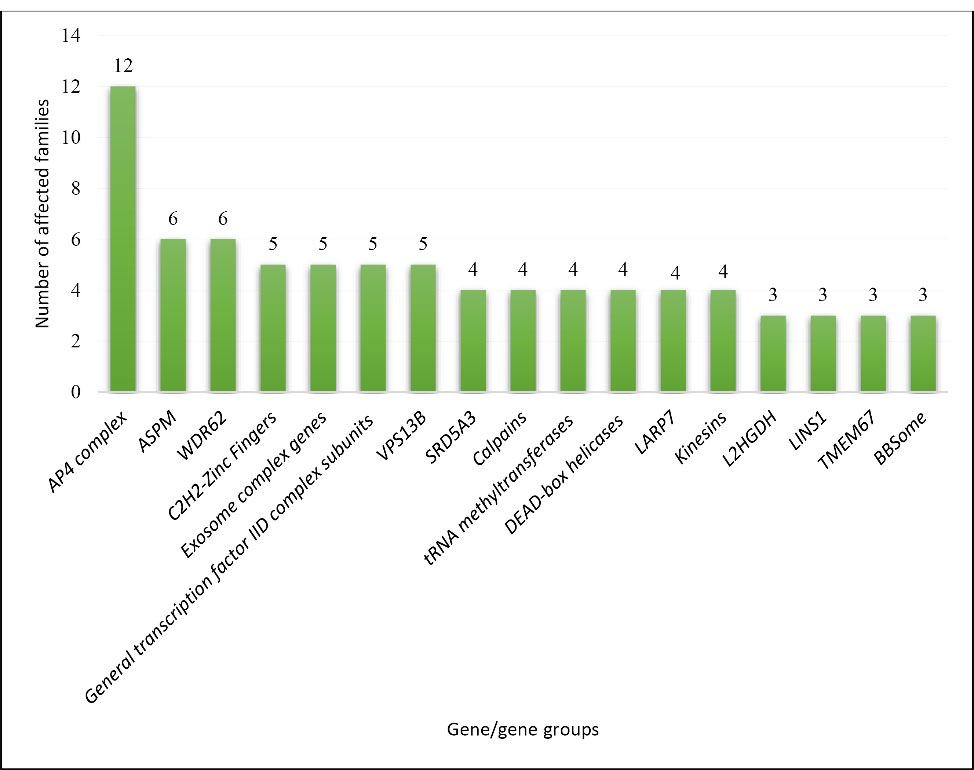
Figure 2.
Number of Families with the Most Common Mutated Genes or Gene Groups in Our Cohort.
.
Number of Families with the Most Common Mutated Genes or Gene Groups in Our Cohort.
Further detailed data regarding the putative function of each gene in the pathogenesis of ID and related phenotypes of each gene/gene group are presented in Table 1 and Table 2. Furthermore, for multiple genes, we found two unrelated affected families, as listed in Table 3.
Table 1.
Functions of the Genes and their Associated Phenotypes
|
Category
|
Function of the Genes and Implicated Phenotypes
|
| Adaptor-related protein complex 4 (AP4) |
The AP4 complex is one of the five members of the Adapter Protein family, which is involved in the post-Golgi pathways in transporting cargo from the trans-Golgi to endosomes and autophagosomal structures.19 This complex consists of four subunits, encoded by AP4B1, AP4E1, AP4M1, and AP4S1. The AP4 complex could be involved in the transportation of various cargoes, including low-density lipoprotein receptor, amyloid precursor protein, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, ATG9A, and δ2 glutamate receptors.20 All which are essential for the proper functioning of the brain.21-26
Mutations in AP4 complex genes cause AP4 deficiency syndrome, which is characterized by intellectual disability, spastic tetraplegia, developmental delay, speech disorder, microcephaly, and inability to walk.19 |
| Abnormal spindle-like, microcephaly-associated; ASPM |
ASPM encodes ASPM, a protein localized at the centrosome of apical neural progenitor cells that is involved in mitotic spindle orientation during embryonic neurogenesis27 and is important for the correct proliferation and differentiation of neural progenitor cells during brain development.28 Mutations inthis gene cause autosomal recessive primary microcephaly 5, characterized by ID, microcephaly, sloping forehead, hypoplasia of the corpus callosum, simplified gyral pattern, and speech problems.29,30 |
| WD repeat-containing protein 62; WDR62 |
WDR62 is involved in spindle dynamics and organization, and is important for the proliferation of neural stem cells.31,32
Mutations in this gene cause autosomal recessive primary microcephaly 2, with or without cortical malformations. These patients show microcephaly, cortical malformations, developmental delays, and seizures.33 |
| Cys2His2 zinc finger (C2H2-ZNF); ZNF335, ZNF526, ZNF804A |
C2H2 zinc-finger proteins are the largest family of human TFs. They play a critical role in the transcriptional regulation of neural stem cells that rise to neurons and glial cells; therefore, proper function of these TFs is crucial for normal brain development.34 |
| Exosome complex (EXOSC); EXOSC2, EXOSC3, EXOSC5 |
The EXOSC gene family includes genes responsible for the formation of the RNA-exosome complex. This complex is vital to RNA processing. It consists of ten conserved subunits, including EXOSC1-3 as non-catalytic cap components, EXOSC4-9 as a non-catalytic core, and DIS3 with both exoribonuclease and endonuclease activity.35-37 Studies on zebrafish have suggested that loss of EXOSC2 would lead to reduced small size; loss of spinal motor neurons and disturbance in EXOSC3 would result in reduced brain size and defects in the development of spinal motor neurons and the cerebellum.38,39 Loss of function of EXOSC5 in zebrafish causes reduced head and eye size as well as edema.40 |
| General transcription factor IID complex subunits (TAF); TAF1, TAF2, TAF6 |
General TFIID is essential for the transcription initiation of RNA polymerase II. TFIID is a complex consisting of a TBP and 13 conserved factors called TAFs.41,42 TAF1 encodes the largest subunit of TFIID, and is involved in early brain development. RT-PCR studies on cells harboring loss of TAF1 showed changes in gene expression of neuronal ion channels.43 TAF2 acts as a stabilizer in binding TFIID to the core promoter.44 TAF6 encodes part of the core of the TFIID complex, and defective TAF6 can alter the assembly of TFIID.45 |
| Vacuolar Protein Sorting 13 Homolog B; VPS13B |
This gene encodes a protein that is important for non-vesicular lipid transport through intracellular membrane contact sites, and disorganizations in lipid constituents of organelle membranes would cause neurological disorders.46 Studies on flies also showed that VPS13B is necessary for the homeostasis of brain proteins.47
Mutations in this gene would result in a well-characterized disorder, Cohen syndrome, with common clinical features, including ID, developmental delay, microcephaly, eye problems, and facial characteristics.48 |
| Steroid 5-alpha reductase family (SRD5A); SRD5A3 |
This gene encodes an enzyme called steroid 5a-reductase type 3, which is vital for N-glycosylation in the endoplasmic reticulum and has a crucial role in catalyzing the conversion of polyprenol to dolichol.49,50
Mutation in this gene causes Kahrizi syndrome with ID, cataracts, coloboma, kyphosis, and coarse facial features in our cohort.51 |
| La Ribonucleoprotein 7 transcriptional regulator; LARP7 |
This gene encodes a transcriptional regulator protein that acts by binding to 7SK RNA and acts as an inhibitor of transcription by RNA polymerase II.52 Knockdown experiments on rats showed that inhibition of LARP7 could inhibit protein synthesis and reduce ribosomes in hippocampal neurons.53 Mutations in this gene cause LARP7 deficiency, characterized by ID, developmental delay, skeletal anomalies, and behavioral problems.54 |
| Calpains (CAPN); CAPN10, CAPN9 |
Calpains are a highly conserved group of calcium-dependent cysteine proteases that regulate synaptic plasticity and programmed neuronal death.55,56 They are essential for early embryo development through nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and Wingless-related integration site (Wnt) pathways.57 |
| tRNA methyltransferases (TRMT); TRMT1, TRMT10A |
Both genes encode tRNA methyltransferases that are involved in various cellular functions. Studies have shown that TRMT10A is highly expressed in the embryonic and fetal brain58 and defective TRMT1 can enhance redox homeostasis. As a result, neural stem cells deteriorate due to higher sensitivity to reactive oxygen species and perturb normal neurogenesis.59 |
| DEAD-box helicases (DDX); DDX3X, DDX50 |
The DEAD-box helicase family is a large family of ATP-dependent RNA helicases with a highly conserved Asp-Glu-Ala-Asp [D-E-A-D] motif that is involved in RNA metabolism.60 Studies have shown that alterations in DDX3X would lead to perturbation of RNA metabolism and alter the development of the brain cortical region.61 |
| Kinesins (KIF); KIF7, KIF11, KIF4A |
Kinesins are evolutionarily conserved motor proteins, important for the development of the brain and nervous system. They are involved in various biological functions, including cell division and intracellular trafficking.62 |
| L-2-hydroxyglutarate dehydrogenase; L2HGDH |
This gene provides L-2-hydroxyglutarate dehydrogenase, a mitochondrial enzyme involved in the conversion of L-2-hydroxyglutarate to 2-ketoglutarate.63 Studies in mice have shown that a defective form of L-2-hydroxyglutarate dehydrogenase leads to white matter abnormalities, neuroinflammation, improper neurogenesis of the hippocampal region, and neurodegeneration.64 Mutations in this gene cause L-2-hydroxyglutaric aciduria, characterized by ID, cerebellar ataxia, epilepsy, speech problems, and an increased amount of L-2-hydroxyglutaric acid in urine, blood, and cerebrospinal fluid.63 |
| Lines Homolog 1; LINS1 |
Mutations in LINS1 deteriorate the proper function of the WNT signaling pathway, which is involved in the development of the central nervous system and affects cell fate determination in neuronal progenitor cells, neuronal migration and polarization, and synaptogenesis.65,66 Mutations in LINS1 lead to intellectual developmental disorder, autosomal recessive 27. |
| Transmembrane Protein 67; TMEM67 |
TMEM67 encodes Meckelin, a transmembrane protein involved in cerebellar development that controls the Wnt/β-catenin signaling pathway.67 During development and differentiation, Meckelin can act as a WNT receptor and is also involved in centrosome migration during ciliogenesis and primary cilium formation.68 Mutations in TMEM67 can cause a variety of ciliopathies, including Meckel syndrome, Joubert syndrome, and COACH syndrome 1.68 Here, we report three families with a mutation in TMEM67 that caused Joubert syndrome 6, which is categorized with ID, hypoplasia of the cerebellar vermis, molar tooth sign, hypotonia, developmental delay, ataxia, and renal problems. |
| BBSome; BBS7, BBS9, BBS4 |
BBSome is an octameric complex involved in protein trafficking of the ciliary membrane and non-ciliary functions, including the localization of receptors in the cell membrane.69,70 This complex is essential for the appropriate functioning of astrocytes in the brain. Studies have shown that disruption of BBSome causes defects in primary cilia and affects the morphology and metabolism of neurons in the brain.71,72 Mutations in the subunits of the BBSome complex cause Bardet-Biedl syndrome, categorized with ID, central obesity, hypogonadism, retinal dystrophy, renal problems, and post-axial polydactyly.73,74 |
TFs, transcription factors; ID, Intellectual disability; TFIID, transcription factor IID; TBP, TATA-binding protein; TAF, TBP-associated factor; RT-PCR, Real-time polymerase chain reaction.
Table 2.
Details of the Families with Common ID Genes in our Cohort
|
Genes and the Categories |
Chromosomal Variant#
|
OMIM Phenotype
|
Mode of Inheritance
|
Ethnicity of the Families
|
| AP4 complex |
AP4B1
|
NC_000001.10:g.114442649dela |
614066 |
AR |
Persian |
| NC_000001.10:g.114441378_114441379dela |
| NC_000001.10:g.114441425T > Ca |
|
AP4E1
|
NC_000015.9:g.51242065_51242066insNNc |
613744 |
AR |
Azeri |
|
AP4M1
|
NC_000007.13:g.99701748G > Ac |
612936 |
AR |
Kurd |
| NC_000007.13:g.99703887A > Cb |
Persian |
| NC_000007.13:g.99700491dela |
Persian |
| NC_000007.13:g.99703627G > Aa |
Turkmen |
| NC_000007.13:g.99701748G > Aa |
Persian |
| NC_000007.13:g.99701748G > Aa |
Persian |
|
AP4S1
|
NC_000014.8:g.31542174C > Ta |
614067 |
AR |
Baluch |
| NC_000014.8:g.31542174C > Ta |
Persian |
| ASPM |
NC_000001.10:g.197111490_197111491delb |
608716 |
AR |
Baluch |
| NC_000001.10:g.197070329_197070330dupa |
Persian |
| NC_000001.10:g.197070283G > Aa |
Azeri |
| NC_000001.10:g.197091611_197091612dela |
Persian |
| NC_000001.10:g.197070599_197070600dela |
Persian |
NC_000001.10:g.197115270C > Gd
NC_000001.10:g.197091611_197091612deld |
Persian |
| WDR62 |
NC_000019.9:g.36575602A > Ga |
604317 |
AR |
Persian |
| NC_000019.9:g.36546051G > Ta |
| NC_000019.9:g.36594088_36594089dela |
| NC_000019.9:g.36582182C > Tc |
| NC_000019.9:g.36594255deld |
| NC_000019.9:36558235G > Ad |
| C2H2-Zinc Finger |
ZNF335
|
NC_000020.10:g.44588870G > Aa |
615095 |
AR |
Persian |
| NC_000020.10:g.44578005A > Ca |
Persian |
|
ZNF526
|
NC_000019.9:g.42730172G > Cc |
619877 |
AR |
Baluch |
| NC_000019.9:g.42729931G > Ac |
Kurd |
|
ZNF804A
|
NC_000002.11:g.185731147G > Ab |
ID |
AR |
Persian |
| Exosome complex |
EXOSC2
|
NC_000009.11:g.133578439G > Tb |
617763 |
AR |
Persian |
| NC_000009.11:g.133578439G > Td |
Persian |
|
EXOSC3
|
NC_000009.11:g.37783990T > Gd |
614678 |
AR |
Persian |
| NC_000009.11:g.37783990T > Gd |
Persian |
|
EXOSC5
|
NC_000019.9:g.41897789G > Aa |
619576 |
AR |
Persian |
| General transcription factor IID complex subunits |
TAF1
|
NC_000023.10:g.70588006C > Gb |
300966 |
XLR |
Persian |
| NC_000023.10:g.70607141A > Ga |
|
TAF2
|
NC_000008.10:g.120795788A > Gc |
615599 |
AR |
Persian |
| NC_000008.10:g.120805628C > Ad |
Kurd |
|
TAF6
|
NC_000007.13:g.99711522A > Ga |
617126 |
AR |
Azeri |
| VPS13B |
NC_000008.10:g.100732719dela |
216550 |
AR |
Persian |
| NC_000008.10:g.100832347_100832380delinsCa |
Persian |
| NC_000008.10:g.100732719dela |
Persian |
| NC_000008.10:g.100832269dela |
Arab |
| NC_000008.10:g.100568867G > Aa |
Persian |
| Steroid 5-alpha reductase family |
SRD5A3
|
NC_000004.11:g.56212560G > Ab |
612713 |
AR |
Persian |
| NC_000004.11:g.56230382A > Gc |
Baluch |
| NC_000004.11:g.56212705_56212706insNc |
Persian |
| NC_000004.11:g.56212707dupf |
Baluch |
|
LARP7 |
NC_000004.11:g.113575316G > Ca |
615071 |
AR |
Persian |
| NC_000004.11:g.113568633C > Tg |
Persian |
| NC_000004.11:g.113578402_113578405delg |
Azeri |
| NC_000004.11:g.113568536_113568537insNc |
Turk |
|
Calpains
|
CAPN10
|
NC_000002.11:g.241530371_241530376insN[15]c |
601283 |
AR |
Persian |
| NC_000002.11:g.241528849T > Aa |
Arab |
| NC_000002.11:g.241530301C > Ta |
Persian |
|
CAPN9
|
NC_000001.10:g.230898426G > Ta |
ID |
AR |
Arab |
| tRNA methyltransferases |
TRMT1
|
NC_000019.9:g.13223781_13223812delc |
618302 |
AR |
Arab |
| NC_000019.9:g.13223781_13223812dela |
Baluch |
| NC_000019.9:g.13220260_13220261dela |
Azeri |
|
TRMT10A
|
NC_000004.11:g.100478552G > Ta |
616033 |
AR |
Persian |
| DEAD-box helicases |
DDX3X
|
NC_000023.10:g.41204441T > Ab |
300958 |
XLR |
Persian |
| NC_000023.10:g.41203594A > Ga |
| NC_000023.10:g.41204491C > Ta |
|
DDX50
|
NC_000010.10:g.70706241_70706264delb |
ID |
AR |
| Kinesins |
KIF11
|
NC_000010.10:g.94366083C > Tb |
152950 |
AD |
Persian |
|
KIF4A
|
NC_000023.10:g.69607097C > Ta |
300923 |
XLR |
Turk |
|
KIF7
|
NC_000015.9:g.90185556C > Tc |
200990 |
AR |
Persian |
| NC_000015.9:g.90195903T > Cb |
Arab |
| L-2-hydroxyglutarate dehydrogenase |
L2HGDH
|
NC_000014.8:g.50750723G > Aa |
236792 |
AR |
Persian |
| NC_000014.8:g.50734532G > Ac |
Persian |
| NC_000014.8:g.50768804A > Td |
Lur |
|
LINS1 |
NC_000015.9:g.101114094_101114097dela |
614340 |
AR |
Persian |
| NC_000015.9:g.101120983dela |
Kurd |
| NC_000015.9:g.101114094_101114097delc |
Persian |
|
TMEM67 |
NC_000008.10:g.94792831A > Gb |
610688 |
AR |
Persian |
| NC_000008.10:g.94792831A > Ga |
| NC_000008.10:g.94792831A > Ga |
| BBSome |
BBS4
|
NC_000015.9:g.73002041_73004648dela |
615982 |
AR |
Persian |
|
BBS7
|
NC_000004.11:g.122754467_122754472delc |
615984 |
AR |
|
BBS9
|
NC_000007.13:g.33397608G > Aa |
615986 |
AR |
ID, intellectual disability; AR, autosomal recessive; XLR, X-linked recessive; AD, autosomal dominant; NA, not assigned.
# Based on GRCh37(hg19).
a These families were first reported in our previous study.1
b These families were first reported in our previous study.8
c These families were first reported in our previous study.9
d These families were first reported in our previous study.35
e Unpublished data.
f The family was first described in a previous paper.75
g These families were first reported in our previous study.10
Table 3.
Genes with a Mutation in Two Unrelated Affected Families
|
Gene
|
Chromosomal variants#
|
OMIM phenotype
|
Ethnicity of the families
|
|
ADGRG1
|
NC_000016.9:g.57695619C > Tb |
606854 |
Persian |
| NC_000016.9:g.57695794G > Aa |
|
AHI1
|
NC_000006.11:g.135778798G > Ac |
608629 |
Persian |
| NC_000006.11:g.135769570C > Tc |
|
AIMP1
|
NC_000004.11:g.107258194G > Cb |
260600 |
Persian |
| NC_000004.11:g.107252964T > Ga |
|
AK1
|
NC_000009.11:g.130630703C > Ta |
612631 |
Persian |
| NC_000009.11:g.130634140G > Aa |
Arab |
|
ALS2
|
NC_000002.11:g.202569196A > Ga |
205100 |
Persian |
| NC_000002.11:g.202619225C > Ta |
Arab |
|
ASNS
|
NC_000007.13:g.97488183A > Cb |
615574 |
Persian |
| NC_000007.13:g.97498245T > Ca |
|
ATP8A2
|
NC_000013.10:g.26125642G > Tb |
615268 |
Persian |
| NC_000013.10:g.26436510G > Ab |
|
ATRX
|
NC_000023.10:g.76855934A > Ga |
309580 |
Persian |
| NC_000023.10:g.76875953C > Ga |
|
B3GALNT2
|
NC_000001.10:g.235643447G > Aa |
615181 |
Azeri |
| NC_000001.10:g.235621957C > Ta |
Persian |
|
CASK
|
NC_000023.10:g.41416344G > Ca |
300422 |
Arab |
| NC_000023.10:g.41519706G > Aa |
Persian |
|
CDK5RAP2
|
NC_000009.11:g.123201968_123201971delb |
604804 |
Persian |
| NC_000009.11:g.123253590_123253593dela |
Baluch |
|
CEP104
|
NC_000001.10:g.3742330_3742331insAAa |
616781 |
Persian |
| NC_000001.10:g.3746500dupa |
Lur |
|
DYM
|
NC_000018.9:g.46889551dela |
223800 |
Persian |
| NC_000018.9:g.46808420G > Aa |
|
ELP2
|
NC_000018.9:g.33736538G > Tc |
617270 |
Azeri |
| NC_000018.9:g.33739953A > Cc |
Turk |
|
ERLIN2
|
NC_000008.10:g.37599315_37599677delinsCTGTGa |
611225 |
Azeri |
| NC_000008.10:g.37595547G > A c |
Persian |
|
GAMT
|
NC_000019.9:g.1398999dela |
612736 |
Persian |
| NC_000019.9:g.1398999dela |
|
GMPPA
|
NC_000002.11:g.220368858C > Ab |
615510 |
Persian |
| NC_000002.11:g.220370723G > Aa |
|
IPP
|
NC_000001.10:g.46179920_46185014dela |
ID |
Persian |
| NC_000001.10:g.46182687C > Ta |
|
ITGAV
|
NC_000002.11:g.187541960_187541962dela |
ID |
Persian |
| NC_000002.11:g.187529348G > Aa |
Arab |
|
MAN1B1
|
NC_000009.11:g.139995540C > Tc |
614202 |
Persian |
| NC_000009.11:g.140001735deld |
Lur |
|
NDST1
|
NC_000005.9:g.149922489G > Ta |
616116 |
Persian |
| NC_000005.9:g.149925029G > Ac |
|
NEURL4
|
NC_000017.10:g.7224505G > Ab |
ID |
Persian |
| NC_000017.10:g.7222392dupa |
|
ORC1
|
NC_000001.10:g.52851591G > Aa |
224690 |
Lur |
| NC_000001.10:g.52850232T > Ca |
Turkmen |
|
PIDD1
|
NC_000011.9:g.800015C > Ta |
ID |
Persian |
| NC_000011.9:g.799846G > Aa |
|
PRKCG
|
NC_000019.9:g.54394928_54396645del c |
605361 |
Persian |
| NC_000019.9:g.54403866G > Tc |
|
PRRT2
|
NC_000016.9:g.29825024dupa |
602066 |
Kurd |
| NC_000016.9:g.29825015_29825016insNc |
|
RDH11
|
NC_000014.8:g.68145040dupa |
616108 |
Persian |
| NC_000014.8:g.68159744T > Cd |
Persian |
|
RNASEH2C
|
NC_000011.9:g.65487533G > Ab |
610329 |
Persian |
| NC_000011.9:g.65487856G > Aa |
Baluch |
|
RNFT2 |
NC_000012.11:g.117274037T > Ca |
ID |
Persian |
| NC_000012.11:g.117274037T > Ca |
|
SCAPER
|
NC_000015.9:g.77064235G > Aa |
618195 |
Baluch |
| NC_000015.9:g.77064240_77064241insNc |
Persian |
|
SUCLA2
|
NC_000013.10:g.48528645G > Ab |
612073 |
Kurd |
| NC_000013.10:g.48562777T > Gb |
Azeri |
|
SURF1
|
NC_000009.11:g.136219373A > Gc |
220110 |
Turk |
| NC_000009.11:g.136218979C > Aa |
Arab |
|
TSEN54
|
NC_000017.10:g.73513639G > Tb |
610204 |
Persian |
| NC_000017.10:g.73513639G > Ta |
Kurd |
|
TTC5
|
NC_000014.8:g.20766998dela |
619244 |
Turk |
| NC_000014.8:g.20774045C > Ta |
Baluch |
|
TWNK |
NC_000010.10:g.102748841C > Aa |
616138 |
Persian |
| NC_000010.10:g.102748841C > Ad |
Persian |
|
UBE3B
|
NC_000012.11:g.109921396G > Ab |
244450 |
Persian |
| NC_000012.11:g.109935697T > Ca |
Arab |
ID, intellectual disability.
# Based on GRCh37(hg19).
a These families were first reported in our previous study.1
b These families were first reported in our previous study.8
c These families were first reported in our previous study.9
d Unpublished data.
Comparison of our Study with Seven Studies Reporting ID Cohorts
We compiled two lists of reported ID genes among seven studies from neighboring countries with a high consanguinity rate that included ID cohorts.12-18 This comparison resulted in the Venn diagram depicted in Figure 3. We also extracted repetitive genes (Supplementary File 1, Table S1) embedded in these three lists and compared them by depicting another Venn diagram shown in Figure 4.76 The details of these comparisons are shown in Table 4. For both of these comparisons, copy number variations were excluded.
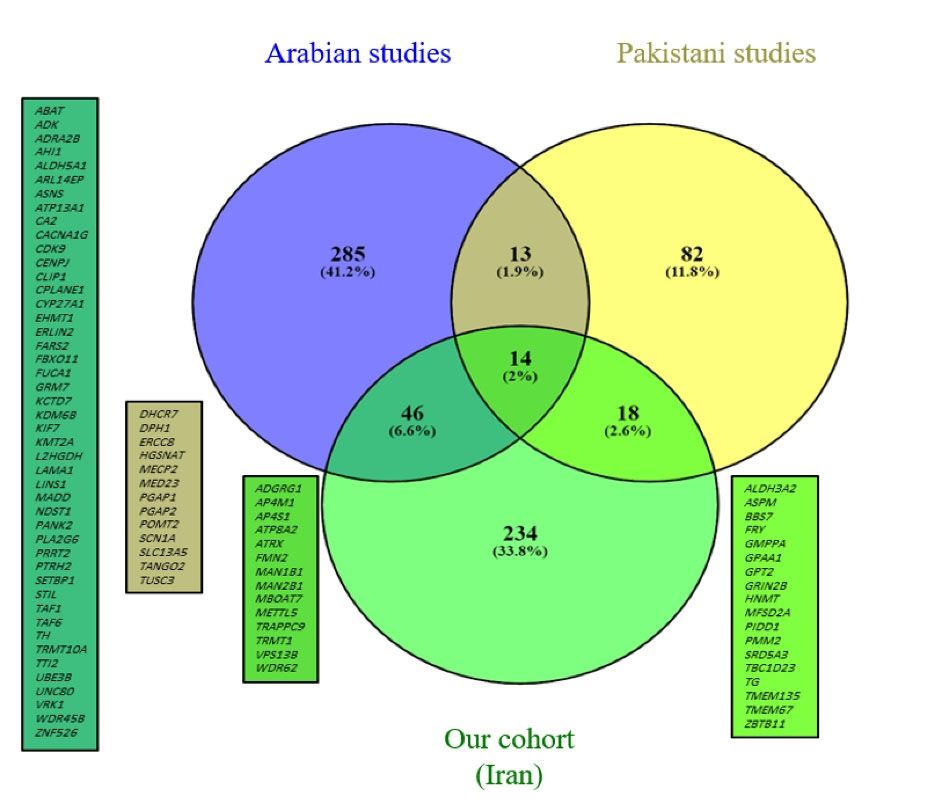
Figure 3.
Venn Diagram Showing ID Genes Reported in our Cohort and the Pakistani and Arab Groups.
.
Venn Diagram Showing ID Genes Reported in our Cohort and the Pakistani and Arab Groups.
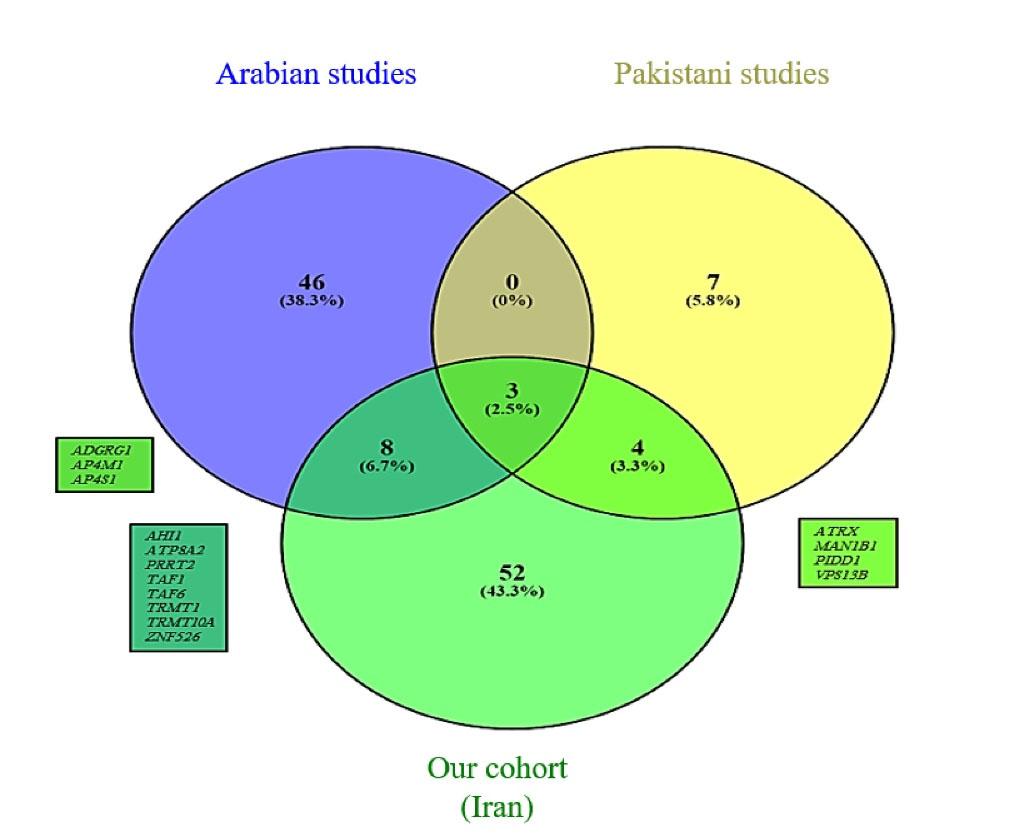
Figure 4.
Venn Diagram Showing Repetitive ID Genes Reported in our Cohort and the Pakistani and Arab Groups.
.
Venn Diagram Showing Repetitive ID Genes Reported in our Cohort and the Pakistani and Arab Groups.
Table 4.
Details of Gene Comparison Between Pakistani and Arab Groups
|
Cohorts/Number of Genes
|
Our Cohort (Iranian Population)
|
Pakistani Groups
|
Arab Groups
|
| Total number of genes |
312 |
127 |
358 |
| Number of repetitive genes in total |
67 |
14 |
57 |
Discussion
Based on an epidemiological study of a large Iranian ID cohort, we were able to categorize the most common ID genes into 17 groups (AP4 complex, ASPM, WDR62, C2H2-Zinc fingers, exosome complex genes, General transcription factor IID subunits, VPS13B, SRD5A3, LARP7, calpain genes, tRNA methyltransferases, kinesins, DEAD-box helicases, L2HGDH, LINS1, TMEM67, and BBSome complex genes). Each group was repeatedly reported for at least three families in our cohort. Because of the high consanguinity rate in our population, 87.87% of these genes demonstrated an AR mode of inheritance. The most common syndromic ID in our study was AP4 deficiency syndrome, which was reported in 12 families and the most common non-syndromic ARID gene was ASPM.
For 36 ID genes, we could identify two unrelated families. For several genes, we found two unrelated families with the same mutations. These included families with (NC_000008.10:g.100732719del, p.Phe2293Leufs*24) in VPS13B, families with (NC_000012.11:g.117274037T > C, p.Cys384Arg) in RNFT2,and families with (NC_000019.9:g.1398999del, p.Gly164Alafs*14) in GAMT. In another study in 2015, Rafiq et al reported (p.Phe2293Leufs*24) in two unrelated Pakistani families of Baloch population.77 On the other hand, for the recurrent variant in TMEM67, Dehghani et al found the same mutation among 12 Iranian nuclear families and suggested the variant as a founder mutation in the Iranian population.78 Our study supports this hypothesis and confirms the prioritization of this variant for the diagnosis of Iranian patients with Joubert syndrome. At the same time, more studies are needed to confirm our hypothesis. Studies have shown that the variant of GAMT has been reported frequently in various families from Turkey, Israeli Arabs, Italy, and Iran.1,79-81 It seems that the glycine at position 164 is a highly conserved amino acid, and a mutation at this position is one of the most prevalent alterations in GAMT.
According to HGMD and ClinVar, worldwide epidemiological studies on ARID showed that only a small number of these genes appear to have frequent variant reports, including GALT, VPS13B, ASPM, SPG11, MUT, GLDC, CEP290, POLG, LAMA2, and SMPD1.6 Two of these genes (VPS13B, ASPM) were also frequent in our cohort. In 2018, Jamra6 estimated that because both these syndromic genes have been well-known for a long time, a large number of reports are available. Although these genes have been known for a long time, our cross-sectional data showed a high prevalence of both genes, suggesting that they are two prevalent ARID genes.
The comparison of ID genes between our Iranian cohort, the Pakistani cohort, and Arab cohorts showed that Iran and Arabs have more common genes in comparison to Pakistani cohort. At this stage, we cannot claim that this similarity in ID genes is due to a more similar genetic background between these two groups of people, and more comprehensive studies are needed. We found 14 genes common between the three groups including ADGRG1, AP4M1, AP4S1, ATP8A2, ATRX, FMN2, MAN1B1, MAN2B1, MBOAT7, METTL5, TRAPPC9, TRMT1, VPS13B, and WDR62. The first three genes (AP4M1 and AP4S1 cause AP4 deficiency syndrome and ADGRG1 causes bilateral frontoparietal polymicrogyria) are repeated among these three groups of people, and they seem to be among the most common ID genes in consanguineous marriages.
Along with much better recognition of the role of genetic factors in ID in recent decades, the gap in epidemiological studies of genetic factors in ID has become more evident, and a large number of genes involved in this phenotype are yet to be discovered. Defining the prevalence of ID-mutated genes in Iran and having accurate statistical data help us make better strategic decisions on genetic and clinical diagnostics of IDs in the Iranian population and prevent the occurrence of such costly disabilities. Due to the large sample size, our data could enhance the design of targeted NGS platforms, mainly population-specific diagnostic tools.
Supplementary Files
Supplementary file 1 contains Table S1.
(xlsx)
Acknowledgements
We are grateful to all patients and families for their participation in this study. This study was supported by Elite Researcher Grant Committee under award number 996149 to Kimia Kahrizi from the National Institute for Medical Research Development (NIMAD), Tehran. Iran.
Competing Interests
The authors declare no conflict of interest.
Ethical Approval
This study was approved by the Ethics Committee of Genetics Research Center, at the University of Social Welfare and Rehabilitation Sciences, Tehran, Iran.
References
- Hu H, Kahrizi K, Musante L, Fattahi Z, Herwig R, Hosseini M. Genetics of intellectual disability in consanguineous families. Mol Psychiatry 2019; 24(7):1027-39. doi: 10.1038/s41380-017-0012-2 [Crossref] [ Google Scholar]
- Maia N, Nabais Sá MJ, Melo-Pires M, de Brouwer APM, Jorge P. Intellectual disability genomics: current state, pitfalls and future challenges. BMC Genomics 2021; 22(1):909. doi: 10.1186/s12864-021-08227-4 [Crossref] [ Google Scholar]
- Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet 2016; 17(1):9-18. doi: 10.1038/nrg3999 [Crossref] [ Google Scholar]
- Kochinke K, Zweier C, Nijhof B, Fenckova M, Cizek P, Honti F. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet 2016; 98(1):149-64. doi: 10.1016/j.ajhg.2015.11.024 [Crossref] [ Google Scholar]
- Iqbal Z, van Bokhoven H. Identifying genes responsible for intellectual disability in consanguineous families. Hum Hered 2014; 77(1-4):150-60. doi: 10.1159/000360539 [Crossref] [ Google Scholar]
- Jamra R. Genetics of autosomal recessive intellectual disability. Med Genet 2018; 30(3):323-7. doi: 10.1007/s11825-018-0209-z [Crossref] [ Google Scholar]
- Musante L, Ropers HH. Genetics of recessive cognitive disorders. Trends Genet 2014; 30(1):32-9. doi: 10.1016/j.tig.2013.09.008 [Crossref] [ Google Scholar]
- Kahrizi K, Hu H, Hosseini M, Kalscheuer VM, Fattahi Z, Beheshtian M. Effect of inbreeding on intellectual disability revisited by trio sequencing. Clin Genet 2019; 95(1):151-9. doi: 10.1111/cge.13463 [Crossref] [ Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011; 478(7367):57-63. doi: 10.1038/nature10423 [Crossref] [ Google Scholar]
- Kazemi G, Peymani F, Mohseni M, Zare Ashrafi F, Arzhangi S, Ardalani F. Novel mutation in LARP7 in two Iranian consanguineous families with syndromic intellectual disability and facial dysmorphism. Arch Iran Med 2020; 23(12):842-7. doi: 10.34172/aim.2020.112 [Crossref] [ Google Scholar]
- Fattahi Z, Beheshtian M, Mohseni M, Poustchi H, Sellars E, Nezhadi SH. Iranome: a catalog of genomic variations in the Iranian population. Hum Mutat 2019; 40(11):1968-84. doi: 10.1002/humu.23880 [Crossref] [ Google Scholar]
- Anazi S, Maddirevula S, Salpietro V, Asi YT, Alsahli S, Alhashem A. Expanding the genetic heterogeneity of intellectual disability. Hum Genet 2017; 136(11-12):1419-29. doi: 10.1007/s00439-017-1843-2 [Crossref] [ Google Scholar]
- Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep 2015; 10(2):148-61. doi: 10.1016/j.celrep.2014.12.015 [Crossref] [ Google Scholar]
- Reuter MS, Tawamie H, Buchert R, Hosny Gebril O, Froukh T, Thiel C. Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry 2017; 74(3):293-9. doi: 10.1001/jamapsychiatry.2016.3798 [Crossref] [ Google Scholar]
- Anazi S, Maddirevula S, Faqeih E, Alsedairy H, Alzahrani F, Shamseldin HE. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry 2017; 22(4):615-24. doi: 10.1038/mp.2016.113 [Crossref] [ Google Scholar]
- Shaheen R, Patel N, Shamseldin H, Alzahrani F, Al-Yamany R, ALMoisheer A. Accelerating matchmaking of novel dysmorphology syndromes through clinical and genomic characterization of a large cohort. Genet Med 2016; 18(7):686-95. doi: 10.1038/gim.2015.147 [Crossref] [ Google Scholar]
- Riazuddin S, Hussain M, Razzaq A, Iqbal Z, Shahzad M, Polla DL. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol Psychiatry 2017; 22(11):1604-14. doi: 10.1038/mp.2016.109 [Crossref] [ Google Scholar]
- Harripaul R, Vasli N, Mikhailov A, Rafiq MA, Mittal K, Windpassinger C. Mapping autosomal recessive intellectual disability: combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol Psychiatry 2018; 23(4):973-84. doi: 10.1038/mp.2017.60 [Crossref] [ Google Scholar]
- Beheshtian M, Akhtarkhavari T, Mehvari S, Mohseni M, Fattahi Z, Abedini SS. Comprehensive genotype-phenotype correlation in AP-4 deficiency syndrome; adding data from a large cohort of Iranian patients. Clin Genet 2021; 99(1):187-92. doi: 10.1111/cge.13845 [Crossref] [ Google Scholar]
- Davies AK, Itzhak DN, Edgar JR, Archuleta TL, Hirst J, Jackson LP. AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A. Nat Commun 2018; 9(1):3958. doi: 10.1038/s41467-018-06172-7 [Crossref] [ Google Scholar]
- Wang SH, Huang Y, Yuan Y, Xia WQ, Wang P, Huang R. LDL receptor knock-out mice show impaired spatial cognition with hippocampal vulnerability to apoptosis and deficits in synapses. Lipids Health Dis 2014; 13:175. doi: 10.1186/1476-511x-13-175 [Crossref] [ Google Scholar]
- Engel DF, Grzyb AN, de Oliveira J, Pötzsch A, Walker TL, Brocardo PS. Impaired adult hippocampal neurogenesis in a mouse model of familial hypercholesterolemia: a role for the LDL receptor and cholesterol metabolism in adult neural precursor cells. Mol Metab 2019; 30:1-15. doi: 10.1016/j.molmet.2019.09.002 [Crossref] [ Google Scholar]
- Lee SH, Kang J, Ho A, Watanabe H, Bolshakov VY, Shen J. APP family regulates neuronal excitability and synaptic plasticity but not neuronal survival. Neuron 2020;108(4):676-90.e8. 10.1016/j.neuron.2020.08.011.
- Pereyra M, Medina JH. AMPA receptors: a key piece in the puzzle of memory retrieval. Front Hum Neurosci 2021; 15:729051. doi: 10.3389/fnhum.2021.729051 [Crossref] [ Google Scholar]
- Yamaguchi J, Suzuki C, Nanao T, Kakuta S, Ozawa K, Tanida I. Atg9a deficiency causes axon-specific lesions including neuronal circuit dysgenesis. Autophagy 2018; 14(5):764-77. doi: 10.1080/15548627.2017.1314897 [Crossref] [ Google Scholar]
- Kohda K, Kakegawa W, Yuzaki M. Unlocking the secrets of the δ2 glutamate receptor: a gatekeeper for synaptic plasticity in the cerebellum. Commun Integr Biol 2013; 6(6):e26466. doi: 10.4161/cib.26466 [Crossref] [ Google Scholar]
- Létard P, Drunat S, Vial Y, Duerinckx S, Ernault A, Amram D. Autosomal recessive primary microcephaly due to ASPM mutations: an update. Hum Mutat 2018; 39(3):319-32. doi: 10.1002/humu.23381 [Crossref] [ Google Scholar]
- Garrett L, Chang YJ, Niedermeier KM, Heermann T, Enard W, Fuchs H. A truncating Aspm allele leads to a complex cognitive phenotype and region-specific reductions in parvalbuminergic neurons. Transl Psychiatry 2020; 10(1):66. doi: 10.1038/s41398-020-0686-0 [Crossref] [ Google Scholar]
- Abdel-Hamid MS, Ismail MF, Darwish HA, Effat LK, Zaki MS, Abdel-Salam GM. Molecular and phenotypic spectrum of ASPM-related primary microcephaly: identification of eight novel mutations. Am J Med Genet A 2016; 170(8):2133-40. doi: 10.1002/ajmg.a.37724 [Crossref] [ Google Scholar]
- Hussain S, Nawaz A, Hamid M, Ullah W, Khan IN, Afshan M. Mutation screening of multiple Pakistani MCPH families revealed novel and recurrent protein-truncating mutations of ASPM. Biotechnol Appl Biochem 2022; 69(6):2296-303. doi: 10.1002/bab.2286 [Crossref] [ Google Scholar]
- Huang J, Liang Z, Guan C, Hua S, Jiang K. WDR62 regulates spindle dynamics as an adaptor protein between TPX2/Aurora A and katanin. J Cell Biol 2021; 220(8):e202007167. doi: 10.1083/jcb.202007167 [Crossref] [ Google Scholar]
- Zhi Y, Zhou X, Yu J, Yuan L, Zhang H, Ng DCH. Pathophysiological significance of WDR62 and JNK signaling in human diseases. Front Cell Dev Biol 2021; 9:640753. doi: 10.3389/fcell.2021.640753 [Crossref] [ Google Scholar]
- Zombor M, Kalmár T, Nagy N, Berényi M, Telcs B, Maróti Z. A novel WDR62 missense mutation in microcephaly with abnormal cortical architecture and review of the literature. J Appl Genet 2019; 60(2):151-62. doi: 10.1007/s13353-019-00486-y [Crossref] [ Google Scholar]
- Al-Naama N, Mackeh R, Kino T. C2H2-type zinc finger proteins in brain development, neurodevelopmental, and other neuropsychiatric disorders: systematic literature-based analysis. Front Neurol 2020; 11:32. doi: 10.3389/fneur.2020.00032 [Crossref] [ Google Scholar]
- Beheshtian M, Fattahi Z, Fadaee M, Vazehan R, Jamali P, Parsimehr E. Identification of disease-causing variants in the EXOSC gene family underlying autosomal recessive intellectual disability in Iranian families. Clin Genet 2019; 95(6):718-25. doi: 10.1111/cge.13549 [Crossref] [ Google Scholar]
- Ulmke PA, Xie Y, Sokpor G, Pham L, Shomroni O, Berulava T. Post-transcriptional regulation by the exosome complex is required for cell survival and forebrain development via repression of P53 signaling. Development 2021; 148(3):dev188276. doi: 10.1242/dev.188276 [Crossref] [ Google Scholar]
- de Amorim J, Slavotinek A, Fasken MB, Corbett AH, Morton DJ. Modeling pathogenic variants in the RNA exosome. RNA Dis 2020; 7:e1166. [ Google Scholar]
- Yatsuka H, Hada K, Shiraishi H, Umeda R, Morisaki I, Urushibata H. Exosc2 deficiency leads to developmental disorders by causing a nucleotide pool imbalance in zebrafish. Biochem Biophys Res Commun 2020; 533(4):1470-6. doi: 10.1016/j.bbrc.2020.10.044 [Crossref] [ Google Scholar]
- Wan J, Yourshaw M, Mamsa H, Rudnik-Schöneborn S, Menezes MP, Hong JE. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet 2012; 44(6):704-8. doi: 10.1038/ng.2254 [Crossref] [ Google Scholar]
- Slavotinek A, Misceo D, Htun S, Mathisen L, Frengen E, Foreman M. Biallelic variants in the RNA exosome gene EXOSC5 are associated with developmental delays, short stature, cerebellar hypoplasia and motor weakness. Hum Mol Genet 2020; 29(13):2218-39. doi: 10.1093/hmg/ddaa108 [Crossref] [ Google Scholar]
- Fant CB, Levandowski CB, Gupta K, Maas ZL, Moir J, Rubin JD, et al. TFIID enables RNA polymerase II promoter-proximal pausing. Mol Cell 2020;78(4):785-93.e8. 10.1016/j.molcel.2020.03.008.
- Patel AB, Louder RK, Greber BJ, Grünberg S, Luo J, Fang J, et al. Structure of human TFIID and mechanism of TBP loading onto promoter DNA. Science 2018;362(6421). 10.1126/science.aau8872.
- Hurst SE, Liktor-Busa E, Moutal A, Parker S, Rice S, Szelinger S. A novel variant in TAF1 affects gene expression and is associated with X-linked TAF1 intellectual disability syndrome. Neuronal Signal 2018; 2(3):NS20180141. doi: 10.1042/ns20180141 [Crossref] [ Google Scholar]
- Lesieur-Sebellin M, Capri Y, Grisval M, Courtin T, Burtz A, Thevenon J. Phenotype associated with TAF2 biallelic mutations: a clinical description of four individuals and review of the literature. Eur J Med Genet 2021; 64(11):104323. doi: 10.1016/j.ejmg.2021.104323 [Crossref] [ Google Scholar]
- El-Saafin F, Curry C, Ye T, Garnier JM, Kolb-Cheynel I, Stierle M. Homozygous TAF8 mutation in a patient with intellectual disability results in undetectable TAF8 protein, but preserved RNA polymerase II transcription. Hum Mol Genet 2018; 27(12):2171-86. doi: 10.1093/hmg/ddy126 [Crossref] [ Google Scholar]
- Dziurdzik SK, Conibear E. The Vps13 family of lipid transporters and its role at membrane contact sites. Int J Mol Sci 2021; 22(6):2905. doi: 10.3390/ijms22062905 [Crossref] [ Google Scholar]
- Vonk JJ, Yeshaw WM, Pinto F, Faber AI, Lahaye LL, Kanon B. Drosophila Vps13 is required for protein homeostasis in the brain. PLoS One 2017; 12(1):e0170106. doi: 10.1371/journal.pone.0170106 [Crossref] [ Google Scholar]
- Momtazmanesh S, Rayzan E, Shahkarami S, Rohlfs M, Klein C, Rezaei N. A novel VPS13B mutation in Cohen syndrome: a case report and review of literature. BMC Med Genet 2020; 21(1):140. doi: 10.1186/s12881-020-01075-1 [Crossref] [ Google Scholar]
- Cantagrel V, Lefeber DJ, Ng BG, Guan Z, Silhavy JL, Bielas SL. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell 2010; 142(2):203-17. doi: 10.1016/j.cell.2010.06.001 [Crossref] [ Google Scholar]
- Leonard H, Wen X. The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev 2002; 8(3):117-34. doi: 10.1002/mrdd.10031 [Crossref] [ Google Scholar]
- Kahrizi K, Najmabadi H, Kariminejad R, Jamali P, Malekpour M, Garshasbi M. An autosomal recessive syndrome of severe mental retardation, cataract, coloboma and kyphosis maps to the pericentromeric region of chromosome 4. Eur J Hum Genet 2009; 17(1):125-8. doi: 10.1038/ejhg.2008.159 [Crossref] [ Google Scholar]
- Hasler D, Meister G, Fischer U. Stabilize and connect: the role of LARP7 in nuclear non-coding RNA metabolism. RNA Biol 2021; 18(2):290-303. doi: 10.1080/15476286.2020.1767952 [Crossref] [ Google Scholar]
- Slomnicki LP, Malinowska A, Kistowski M, Palusinski A, Zheng JJ, Sepp M. Nucleolar enrichment of brain proteins with critical roles in human neurodevelopment. Mol Cell Proteomics 2016; 15(6):2055-75. doi: 10.1074/mcp.M115.051920 [Crossref] [ Google Scholar]
- Imbert-Bouteille M, Mau Them FT, Thevenon J, Guignard T, Gatinois V, Riviere JB. LARP7 variants and further delineation of the Alazami syndrome phenotypic spectrum among primordial dwarfisms: 2 sisters. Eur J Med Genet 2019; 62(3):161-6. doi: 10.1016/j.ejmg.2018.07.003 [Crossref] [ Google Scholar]
- Briz V, Baudry M. Calpains: master regulators of synaptic plasticity. Neuroscientist 2017; 23(3):221-31. doi: 10.1177/1073858416649178 [Crossref] [ Google Scholar]
- Cheng SY, Wang SC, Lei M, Wang Z, Xiong K. Regulatory role of calpain in neuronal death. Neural Regen Res 2018; 13(3):556-62. doi: 10.4103/1673-5374.228762 [Crossref] [ Google Scholar]
- Araujo H, Julio A, Cardoso M. Translating genetic, biochemical and structural information to the calpain view of development. Mech Dev 2018; 154:240-50. doi: 10.1016/j.mod.2018.07.011 [Crossref] [ Google Scholar]
- Swinehart WE, Jackman JE. Diversity in mechanism and function of tRNA methyltransferases. RNA Biol 2015; 12(4):398-411. doi: 10.1080/15476286.2015.1008358 [Crossref] [ Google Scholar]
- Dewe JM, Fuller BL, Lentini JM, Kellner SM, Fu D. TRMT1-catalyzed tRNA modifications are required for redox homeostasis to ensure proper cellular proliferation and oxidative stress survival. Mol Cell Biol 2017; 37(21):e00214-17. doi: 10.1128/mcb.00214-17 [Crossref] [ Google Scholar]
- Mo J, Liang H, Su C, Li P, Chen J, Zhang B. DDX3X: structure, physiologic functions and cancer. Mol Cancer 2021; 20(1):38. doi: 10.1186/s12943-021-01325-7 [Crossref] [ Google Scholar]
- Lennox AL, Hoye ML, Jiang R, Johnson-Kerner BL, Suit LA, Venkataramanan S, et al. Pathogenic DDX3X mutations impair RNA metabolism and neurogenesis during fetal cortical development. Neuron 2020;106(3):404-20.e8. 10.1016/j.neuron.2020.01.042.
- Konjikusic MJ, Gray RS, Wallingford JB. The developmental biology of kinesins. Dev Biol 2021; 469:26-36. doi: 10.1016/j.ydbio.2020.09.009 [Crossref] [ Google Scholar]
- Muzammal M, Di Cerbo A, Almusalami EM, Farid A, Khan MA, Ghazanfar S. In silico analysis of the L-2-hydroxyglutarate dehydrogenase gene mutations and their biological impact on disease etiology. Genes (Basel) 2022; 13(4):698. doi: 10.3390/genes13040698 [Crossref] [ Google Scholar]
- Ma S, Sun R, Jiang B, Gao J, Deng W, Liu P, et al. L2HGDH deficiency accumulates L-2-hydroxyglutarate with progressive leukoencephalopathy and neurodegeneration. Mol Cell Biol 2017;37(8). 10.1128/mcb.00492-16.
- Neuhofer CM, Catarino CB, Schmidt H, Seelos K, Alhaddad B, Haack TB. LINS1-associated neurodevelopmental disorder: family with novel mutation expands the phenotypic spectrum. Neurol Genet 2020; 6(5):e500. doi: 10.1212/nxg.0000000000000500 [Crossref] [ Google Scholar]
- Muthusamy B, Bellad A, Prasad P, Bandari AK, Bhuvanalakshmi G, Kiragasur RM. A novel LINS1 truncating mutation in autosomal recessive nonsyndromic intellectual disability. Front Psychiatry 2020; 11:354. doi: 10.3389/fpsyt.2020.00354 [Crossref] [ Google Scholar]
- Bui TPH, Nguyen NT, Ngo VD, Nguyen HN, Ly TTH, Do HD. Novel compound heterozygous TMEM67 variants in a Vietnamese family with Joubert syndrome: a case report. BMC Med Genet 2020; 21(1):18. doi: 10.1186/s12881-020-0962-0 [Crossref] [ Google Scholar]
- Liu D, Qian D, Shen H, Gong D. Structure of the human Meckel-Gruber protein Meckelin. Sci Adv 2021; 7(45):eabj9748. doi: 10.1126/sciadv.abj9748 [Crossref] [ Google Scholar]
- Singh SK, Gui M, Koh F, Yip MC, Brown A. Structure and activation mechanism of the BBSome membrane protein trafficking complex. Elife 2020; 9:e53322. doi: 10.7554/eLife.53322 [Crossref] [ Google Scholar]
- Zhao Y, Rahmouni K. BBSome: a new player in hypertension and other cardiovascular risks. Hypertension 2022; 79(2):303-13. doi: 10.1161/hypertensionaha.121.17946 [Crossref] [ Google Scholar]
- Singh M, Garrison JE, Wang K, Sheffield VC. Absence of BBSome function leads to astrocyte reactivity in the brain. Mol Brain 2019; 12(1):48. doi: 10.1186/s13041-019-0466-z [Crossref] [ Google Scholar]
- Blaess S, Wachten D. The BBSome: a nexus controlling energy metabolism in the brain. J Clin Invest 2021; 131(8):e148903. doi: 10.1172/jci148903 [Crossref] [ Google Scholar]
- Florea L, Caba L, Gorduza EV. Bardet-Biedl syndrome-multiple kaleidoscope images: insight into mechanisms of genotype-phenotype correlations. Genes (Basel) 2021; 12(9):1353. doi: 10.3390/genes12091353 [Crossref] [ Google Scholar]
- Tsegaw A, Teshome T. Bardet-Biedl syndrome in an Ethiopian. Int Med Case Rep J 2021; 14:177-81. doi: 10.2147/imcrj.s299421 [Crossref] [ Google Scholar]
- Kahrizi K, Hu CH, Garshasbi M, Abedini SS, Ghadami S, Kariminejad R. Next generation sequencing in a family with autosomal recessive Kahrizi syndrome (OMIM 612713) reveals a homozygous frameshift mutation in SRD5A3. Eur J Hum Genet 2011; 19(1):115-7. doi: 10.1038/ejhg.2010.132 [Crossref] [ Google Scholar]
- Oliveros JC. (2007-2015) Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. https://bioinfogp.cnb.csic.es/tools/venny/index.html.
- Rafiq MA, Leblond CS, Saqib MA, Vincent AK, Ambalavanan A, Khan FS. Novel VPS13B mutations in three large Pakistani Cohen syndrome families suggests a Baloch variant with autistic-like features. BMC Med Genet 2015; 16:41. doi: 10.1186/s12881-015-0183-0 [Crossref] [ Google Scholar]
- Dehghani M, Mojarad M, Ghayoor Karimiani E, Vahidi Mehrjardi MY, Sahebalzamani A, Ashrafzadeh F. A common ancestral Asn242Ser mutation in TMEM67 identified in multiple Iranian families with Joubert syndrome. Public Health Genomics 2017; 20(3):188-93. doi: 10.1159/000477560 [Crossref] [ Google Scholar]
- Item CB, Mercimek-Mahmutoglu S, Battini R, Edlinger-Horvat C, Stromberger C, Bodamer O. Characterization of seven novel mutations in seven patients with GAMT deficiency. Hum Mutat 2004; 23(5):524. doi: 10.1002/humu.9238 [Crossref] [ Google Scholar]
- Hengel H, Buchert R, Sturm M, Haack TB, Schelling Y, Mahajnah M. First-line exome sequencing in Palestinian and Israeli Arabs with neurological disorders is efficient and facilitates disease gene discovery. Eur J Hum Genet 2020; 28(8):1034-43. doi: 10.1038/s41431-020-0609-9 [Crossref] [ Google Scholar]
- Mercimek-Mahmutoglu S, Stoeckler-Ipsiroglu S, Adami A, Appleton R, Araújo HC, Duran M. GAMT deficiency: features, treatment, and outcome in an inborn error of creatine synthesis. Neurology 2006; 67(3):480-4. doi: 10.1212/01.wnl.0000234852.43688.bf [Crossref] [ Google Scholar]