Arch Iran Med. 26(3):176-180.
doi: 10.34172/aim.2023.27
Case Report
An Extended Iranian Family with Autosomal Dominant Non-syndromic Hearing Loss Associated with A Nonsense Mutation in the DIAPH1 Gene
Marzieh Mohseni Conceptualization, Data curation, Formal analysis, Investigation, Resources, Software, Validation, Visualization, Writing – original draft, 1, # 
Yusuf Mohammadi Methodology, Writing – original draft, 1, #
Farzane Zare Ashrafi Data curation, Investigation, Resources, Software, Writing – original draft, 1
Fatemeh Ghodratpour Data curation, Methodology, 1
Khadijeh Jalalvand Data curation, 1
Sanaz Arzhangi Data curation, Resources, 1
Mojgan Babanejad Data curation, Resources, 1
Mohammad Hossein Azizi Validation, Writing – review & editing, 2
Kimia Kahrizi Conceptualization, Supervision, Visualization, 1
Hossein Najmabadi Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Visualization, Writing – review & editing, 1, *
Author information:
1Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran
2Associate Professor of Otolaryngology, Academy of Medical Sciences of IR Iran, Tehran, Iran
#Marzieh Mohseni and Yusuf Mohammadi contributed equally to this manuscript.
Abstract
Genetic analysis of non-syndromic hearing loss (NSHL) has been challenged due to marked clinical and genetic heterogeneity. Today, advanced next-generation sequencing (NGS) technologies, such as exome sequencing (ES), have drastically increased the efficacy of gene identification in heterogeneous Mendelian disorders. Here, we present the utility of ES and re-evaluate the phenotypic data for identifying candidate causal variants for previously unexplained progressive moderate to severe NSHL in an extended Iranian family. Using this method, we identified a known heterozygous nonsense variant in exon 26 of the DIAPH1 gene (MIM: 602121), which led to "Deafness, autosomal dominant 1, with or without thrombocytopenia; DFNA1" (MIM: 124900) in this large family in the absence of GJB2 disease-causing variants and also OtoSCOPE-negative results. To the best of our knowledge, this nonsense variant (NM_001079812.3):c.3610C>T (p.Arg1204Ter) is the first report of the DIAPH1 gene variant for autosomal dominant non-syndromic hearing loss (ADNSHL) in Iran.
Keywords: DIAPH1, Exome sequencing, Iran, Non syndromic hearing loss
Copyright and License Information
© 2023 The Author(s).
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article as: Mohseni M, Mohammadi Y, Zare Ashrafi F, Ghodratpour F, Jalalvand K, Arzhangi S, et al. An extended Iranian family with autosomal dominant non-syndromic hearing loss associated with a nonsense mutation in the DIAPH1 gene. Arch Iran Med. 2023;26(3):176-180. doi: 10.34172/aim.2023.27
Introduction
Hearing loss (HL) is the most common neurosensory disorder in humans. The incidence rate of congenital HL is estimated to be around 1–2/1000 and 3–4/1000 newborns in developed and developing countries like Iran, respectively.1 Non-syndromic hearing loss (NSHL) is highly heterogeneous and accounts for about 70% of hereditary HL.1,2 About 20% of NSHL cases have an autosomal dominant mode of inheritance,1 and to date, among more than 120 genes identified for NSHL, 51 genes have been assigned to ADNSHL (https://hereditaryhearingloss.org/). From the past to the present, considering the location of Iran in the consanguineous marriage belt and the plentitude of the population with heterogeneous autosomal recessive disorders, many types of research with different techniques have been performed to disclose the genetic etiology of HL in Iran.1 In recent years, high throughput sequencing technologies, such as exome sequencing (ES), have drastically increased the efficacy of gene identification in heterogeneous Mendelian disorders such as NSHL, of which the most common genes were GJB2, SLC26A4, MYO15A, MYO7A, CDH23, and TMC1 in Iran.1-3
In this report, we describe the first ADNSHL Iranian family with the known DIAPH1 (NM_001079812.3):c.3610C > T (p.Arg1204Ter) mutation using the ES technique. The introduction of this family and the process of predicting the disease-causing variant emphasize the importance of genetic counselling for otolaryngologists, careful clinical data collection, and underlying late-onset occurrence in heterogeneous disorders.
Case Report
Nearly 20 years ago, an extended consanguineous Iranian family from Semnan, northern Iran, with sensorineural HL and apparently an autosomal recessive pedigree, was referred to the Genetics Research Center (GRC) of the University of Social Welfare and Rehabilitation Sciences (USWR), Tehran, Iran, for unravelling the etiology of HL (Figure 1). After obtaining informed consent, clinical evaluation and family history were completed. The affected members evaluated for physical examination and clinical otologic findings were normal; also, no thrombocytopenia was detected. Three patients were examined for HL using pure-tone audiometry screening and showed progressive moderate to severe NSHL (Supplementary File 1).
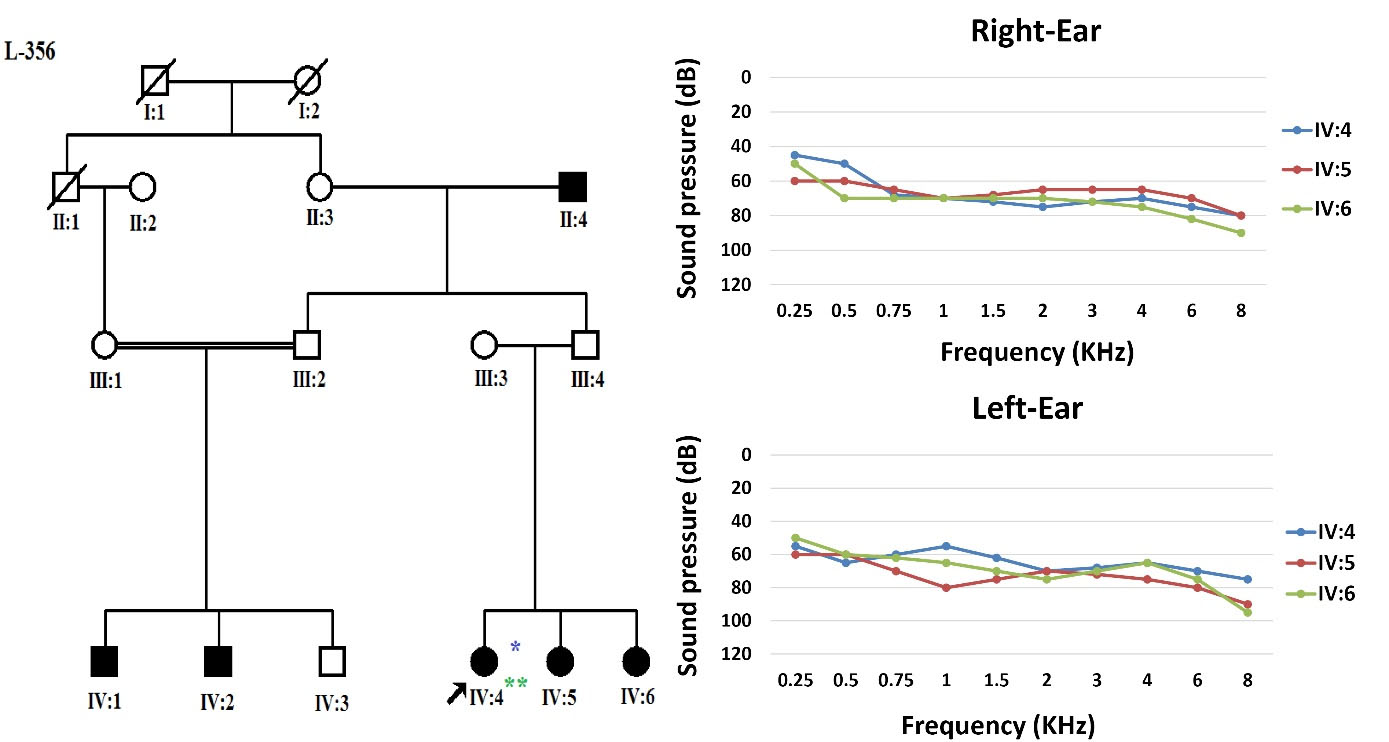
Figure 1.
Previous Pedigree of the Family and Pure Tone Audiometry for Left and Right Ears of Patients (*individual run on OtoSCOPE, **individual run on exome sequencing)
.
Previous Pedigree of the Family and Pure Tone Audiometry for Left and Right Ears of Patients (*individual run on OtoSCOPE, **individual run on exome sequencing)
After sampling the peripheral blood of all 13 members of the family, the DNA of the individuals was extracted by standard salting-out protocols.4 The DNA quantity and quality were accurately assessed by NanoDropTM 2000/2000c Spectrophotometers - Thermo Fisher and running on 1.2% agarose gel.
As a first step, GJB2 screening was performed using Sanger sequencing. Due to the negative results of GJB2 pathogenic variants, the OtoSCOPE panel (V5&V6),5 which covers 89 known deafness-associated genes, including the DIAPH1 gene, was used, and the family was OtoSCOPE-negative for ARNSHL. A few years later, the family was re-evaluated by ES (Illumina NextSeq500 using the SureSelectXT Human All Exon V6 kit; Illumina, Inc.). Tertiary analysis was performed with greater focus on homozygous variants and filtering according to the autosomal recessive model. Variant filtering and prioritization were applied using the ACMG guidelines for the interpretation of sequence variants and the following databases and in-silico algorithms: gnomAD,6 1000 genomes, Iranome,7 OMIM, ClinVar, dbSNP, ExAC Gene Constraints, VS-PolyPhen2, GERP + + , VS-SIFT, and PhyloP. Bioinformatics analysis failed to find a promising homozygous variant in our proband (IV: 4), and just a known heterozygous nonsense variant c.3610C > T in exon 26 of the DIAPH1 gene was identified that was previously reported as a cause of ADNSHL (Table 1). This prompted us to re-evaluate the family and clinical history. After thorough evaluation, it was revealed that individuals (III: 2) and (III: 4) showed late-onset HL (Figure 2). Confirmation of the detected variant and co-segregation study in the family was performed via Sanger sequencing, resulting in the identification of heterozygous DIAPH1 (NM_001079812.3):c.3610C > T (p.Arg1204Ter) mutation as the cause of NSHL in the family (Figure 3). The primers used for sequencing of the candidate variant were: Forward primer: 5’-GTGGGAGAGGGGAAATCAAG-3’, Reverse primer: 5’-AACTCAAATCCCTGGGTCCT-3’.
Table 1.
In Silico Prediction of Variant Identified in DIAPH1 Gene
|
Software
|
Mutation taster
|
phyloP20way_mammalian
|
FATHMM
|
LRT
|
CADD-phred
|
phastCons20way_mammalian
|
DANN
|
CADD-raw
|
ACMG Criteria
|
| Prediction |
Disease causing |
— |
Damaging |
Deleterious |
— |
— |
— |
— |
Pathogenic |
| Score |
0.81 |
1.048 |
0.904 |
0.000012 |
43 |
0.995 |
0.997 |
13.753 |
PVS1-PM2 |
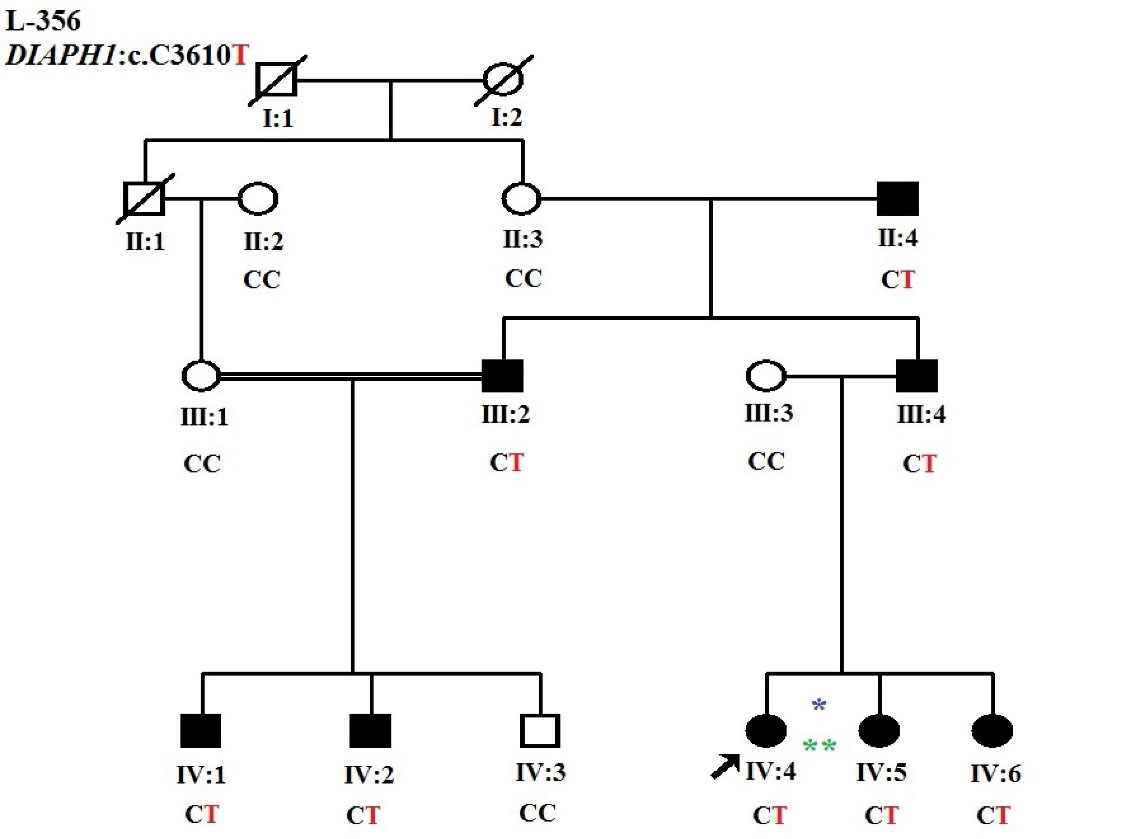
Figure 2.
Updated Pedigree with Autosomal Dominant Inheritance (*individual run on OtoSCOPE, **individual run on exome sequencing)
.
Updated Pedigree with Autosomal Dominant Inheritance (*individual run on OtoSCOPE, **individual run on exome sequencing)
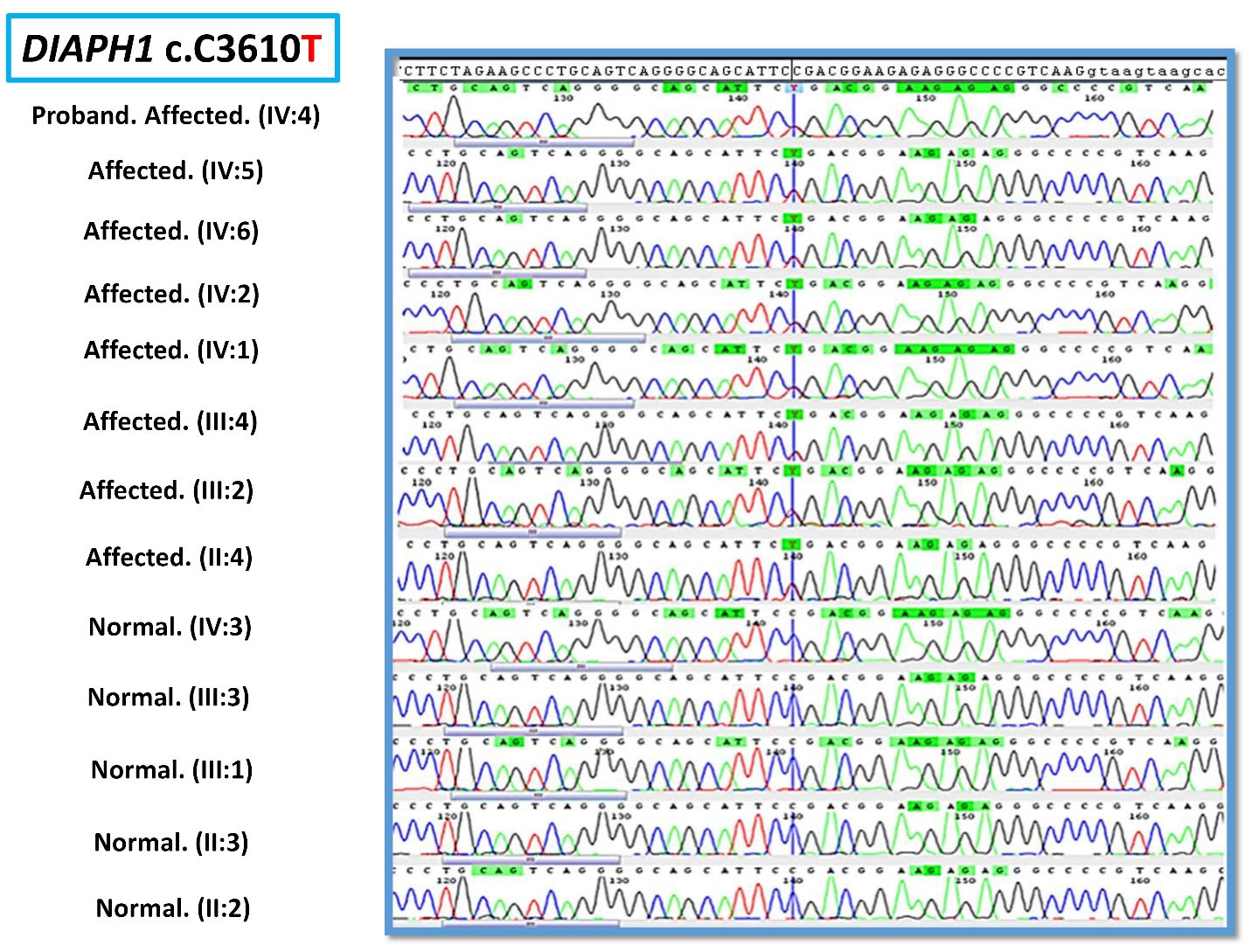
Figure 3.
Electropherogram of c.3610C > T in DIAPH1 Gene in the Family
.
Electropherogram of c.3610C > T in DIAPH1 Gene in the Family
Discussion
The human diaphanous related formin 1; DIAPH1 (also known as DFNA1, DIA1), located on 5q31.3, which encodes homodimeric “Protein diaphanous homolog 1” belongs to the human formin family.8 This protein includes GBD/FH3 (DID), FH1, FH2 and DAD domains and is involved in actin polymerization and microtubule stability (Figure 4).8 Mutations in DIAPH1 are associated with “Deafness, autosomal dominant 1, with or without thrombocytopenia; DFNA1” (MIM: 124900) and “Seizures, cortical blindness, microcephaly syndrome; SCBMS” (MIM: 616632).8
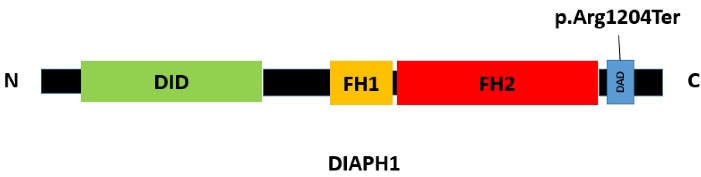
Figure 4.
Schematic Illustration of DIAPH1 Protein
.
Schematic Illustration of DIAPH1 Protein
DIAPH1 plays an essential role in hearing via regulating actin filaments assembly in hair cells in the inner ear of humans. Since identifying DIAPH1 as the first causative gene of ADNSHL in a large Costa Rican family in 1997, among the reported ones, only a few mutations in this gene have been reported with DFNA1 without thrombocytopenia, and most of them have additional symptoms (DFNA1 + Thrombocytopenia).8
Until now, there has been no report of DIAPH1 mutations with ADNSHL in any Iranian family. There are only two articles that have been published on the DIAPH1 gene in Iran; one is the introduction of a rare case with homozygous DIAPH1 mutation leading to SCBMS,9 and the other surveyed the expression patterns of DIAPH1 and virulence genes of dental pathogenic bacteria in oral squamous cell carcinoma patients.10
Here, we report the first Iranian family with ADNSHL and known nonsense (p.Arg1204Ter) mutation located in the DAD domain near the carboxyl terminus of the DIAPH1 protein. This mutation was first reported by Ueyama et al in two unrelated Japanese families suffering from HL in 2016.11 The DIAPH1 (NM_001079812.3):c.3610C > T (p.Arg1204Ter) mutation leads to early termination preceding the (RRKR1204-1207) amino acid motif in the DAD C-terminus and producing constitutively active DIA1 mutant which disrupts the autoinhibitory interaction of DID-DAD domains, and as a result of the continuous activity of the mutant protein, DFNA1 occurs.11
Some reports have suggested that this mutation causes macrothrombocytopenia and autosomal dominant HL through a dominant gain of function mechanism,8,12-14 but the affected individuals in our study showed only ADSNHL without any additional symptoms which would be the confirmation of the previous report of ADSNHL in the DIAPH1.11
Our procedure of genetic counselling and the puzzling situation in the interpretation of the mode of inheritance of the disease in this family highlights the importance of obtaining accurate information in genetic counselling, which is the first and essential step of diagnosis in each medical procedure. This is the first report of DIAPH1 (NM_001079812.3):c.3610C > T (p.Arg1204Ter) variant from Iran. This study could be evidence of pathogenicity for the current variant, in addition to the Japanese research.11
Supplementary File
Supplementary file 1. Pure tone audiometry for the left and right ears of three patients in this family.
(pdf)
Acknowledgements
We thank all patients and their families who participated in this study.
Competing Interests
The authors declare no conflict of interest.
Ethical Approval
We confirm that all authors have no competing interests and followed the ethical issues during this study. This study obtained ethical approval (Institutional ethical approval number: IR.USWR.REC.1396.330) and consent forms were obtained.
References
- Babanejad M, Beheshtian M, Jamshidi F, Mohseni M, Booth KT, Kahrizi K. Genetic etiology of hearing loss in Iran. Hum Genet 2022; 141(3-4):623-31. doi: 10.1007/s00439-021-02421-w [Crossref] [ Google Scholar]
- Mahdieh N, Rabbani B, Wiley S, Akbari MT, Zeinali S. Genetic causes of nonsyndromic hearing loss in Iran in comparison with other populations. J Hum Genet 2010; 55(10):639-48. doi: 10.1038/jhg.2010.96 [Crossref] [ Google Scholar]
- Beheshtian M, Babanejad M, Azaiez H, Bazazzadegan N, Kolbe D, Sloan-Heggen C. Heterogeneity of hereditary hearing loss in Iran: a comprehensive review. Arch Iran Med 2016; 19(10):720-8. [ Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16(3):1215. doi: 10.1093/nar/16.3.1215 [Crossref] [ Google Scholar]
- Sloan-Heggen CM, Babanejad M, Beheshtian M, Simpson AC, Booth KT, Ardalani F. Characterising the spectrum of autosomal recessive hereditary hearing loss in Iran. J Med Genet 2015; 52(12):823-9. doi: 10.1136/jmedgenet-2015-103389 [Crossref] [ Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536(7616):285-91. doi: 10.1038/nature19057 [Crossref] [ Google Scholar]
- Fattahi Z, Beheshtian M, Mohseni M, Poustchi H, Sellars E, Nezhadi SH. Iranome: a catalog of genomic variations in the Iranian population. Hum Mutat 2019; 40(11):1968-84. doi: 10.1002/humu.23880 [Crossref] [ Google Scholar]
- Labat-de-Hoz L, Alonso MA. Formins in human disease. Cells 2021; 10(10):2554. doi: 10.3390/cells10102554 [Crossref] [ Google Scholar]
- Esmaeilzadeh H, Noeiaghdam R, Johari L, Hosseini SA, Nabavizadeh SH, Alyasin SS. Homozygous autosomal recessive DIAPH1 mutation associated with central nervous system involvement and aspergillosis: a rare case. Case Rep Genet 2022; 2022:4142214. doi: 10.1155/2022/4142214 [Crossref] [ Google Scholar]
- Moghimi M, Bakhtiari R, Mehrabadi JF, Jamshidi N, Jamshidi N, Siyadatpanah A. Interaction of human oral cancer and the expression of virulence genes of dental pathogenic bacteria. Microb Pathog 2020; 149:104464. doi: 10.1016/j.micpath.2020.104464 [Crossref] [ Google Scholar]
- Ueyama T, Ninoyu Y, Nishio SY, Miyoshi T, Torii H, Nishimura K. Constitutive activation of DIA1 (DIAPH1) via C-terminal truncation causes human sensorineural hearing loss. EMBO Mol Med 2016; 8(11):1310-24. doi: 10.15252/emmm.201606609 [Crossref] [ Google Scholar]
- Stritt S, Nurden P, Turro E, Greene D, Jansen SB, Westbury SK. A gain-of-function variant in DIAPH1 causes dominant macrothrombocytopenia and hearing loss. Blood 2016; 127(23):2903-14. doi: 10.1182/blood-2015-10-675629 [Crossref] [ Google Scholar]
- Neuhaus C, Lang-Roth R, Zimmermann U, Heller R, Eisenberger T, Weikert M. Extension of the clinical and molecular phenotype of DIAPH1-associated autosomal dominant hearing loss (DFNA1). Clin Genet 2017; 91(6):892-901. doi: 10.1111/cge.12915 [Crossref] [ Google Scholar]
- Ganaha A, Kaname T, Shinjou A, Chinen Y, Yanagi K, Higa T. Progressive macrothrombocytopenia and hearing loss in a large family with DIAPH1 related disease. Am J Med Genet A 2017; 173(10):2826-30. doi: 10.1002/ajmg.a.38411 [Crossref] [ Google Scholar]