Arch Iran Med. 26(12):712-716.
doi: 10.34172/aim.2023.105
Case Report
Purine Nucleoside Phosphorylase Deficiency in Two Unrelated Patients with Autoimmune Hemolytic Anemia and Eosinophilia: Two Novel Mutations
Zahra Alizadeh Conceptualization, Data curation, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing, 1, 2, # 
Mohsen Badalzadeh Conceptualization, Data curation, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing, 1, 2, #
Hanieh Heydarlou Formal analysis, Visualization, Writing – review & editing, 1, 2
Leila Shakerian Formal analysis, Writing – review & editing, 1, 2
Maryam Mahlooji rad Investigation, Resources, Writing – review & editing, 1, 2
Fariborz Zandieh Investigation, Resources, Writing – review & editing, 3
Mohammad Reza Fazlollahi Conceptualization, Data curation, Supervision, Validation, Writing – review & editing, 1, 2, *
Author information:
1Immunology, Asthma and Allergy Research Institute, Tehran University of Medical Sciences, Tehran, Iran
2Children’s Medical Center, Pediatrics Center of Excellence, Tehran University of Medical Sciences, Tehran, Iran
3Department of Asthma, Allergy and Immunology, Bahrami Children Hospital, Tehran University of Medical Sciences, Tehran, Iran
#Contributed equally as first authors.
Abstract
Two Iranian patients with purine nucleoside phosphorylase (PNP) deficiency are described in terms of their clinical and molecular evaluations. PNP deficiency is a rare form of combined immunodeficiency with a profound cellular defect. Patients with PNP deficiency suffer from variable recurrent infections, hypouricemia, and neurological manifestations. Furthermore, patient 1 developed mild cortical atrophy, and patient 2 presented developmental delay, general muscular hypotonia, and food allergy. The two unrelated patients with developed autoimmune hemolytic anemia and T cells lymphopenia and eosinophilia were referred to Immunology, Asthma and Allergy Research Institute (IAARI) in 2019. After taking blood and DNA extraction, genetic analysis of patient 1 was performed by PCR and direct sequencing and whole exome sequencing was applied for patient 2 and the result was confirmed by direct sequencing in the patient and his parents. The genetic result showed two novel variants in exon 3 (c.246_285+9del) and exon 5 (c.569G>T) PNP (NM_000270.4) in the patients, respectively. These variants are considered likely pathogenic based on the American College of Medical Genetics and Genomics (ACMG) guideline. PNP deficiency has a poor prognosis; therefore, early diagnosis would be vital to receive hematopoietic stem cell transplantation (HSCT) as a prominent and successful treatment.
Keywords: Immunodeficiency, Novel mutations, Purine nucleoside phosphorylase, Purine nucleoside phosphorylase deficiency
Copyright and License Information
© 2023 The Author(s).
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article as: Alizadeh Z, Badalzadeh M, Heydarlou H, Shakerian L, Mahlooji rad M, Zandieh F, et al. Purine nucleoside phosphorylase deficiency in two unrelated patients with autoimmune hemolytic anemia and eosinophilia: two novel mutations. Arch Iran Med. 2023;26(12):712-716. doi: 10.34172/aim.2023.105
Introduction
Combined immunodeficiency disease caused by purine nucleoside phosphorylase (PNP) deficiency (OMIM# 613179) is a rare autosomal recessive genetic disorder that severely impairs the function of T cells. Despite this, normal B cell function and number have been reported.1 Patients with PNP deficiency usually suffer from severe infections, failure to thrive, hypouricemia, malignancies, and neutropenia. Neurologic manifestations are present in about two-thirds of the patients and include hypotonia, developmental delay, mental retardation, and ataxia.2 One-third of these patients are diagnosed with autoimmune hemolytic anemia and immune thrombocytopenia.3
PNP deficiency has been reported in about 4% of the patients with severe T cell deficiencies.4 PNP is known as an essential enzyme in the purine salvage pathway that reversibly converts inosine to hypoxanthine and guanosine to guanine.5 PNP-deficient patients have high levels of deoxyguanosine, guanosine and decreased concentrations of guanine and uric acid in their plasma and urine.6 Deoxyguanosine triphosphate (dGTP) accumulation in the mitochondria is toxic to T lymphocytes, inhibits mitochondrial DNA repair, and increases T lymphocyte apoptosis during the selection in thymus.7,8
The neurological dysfunction experienced by patients is caused by the insufficient presence of dGTP in brain tissue, as well as the accumulation of purines within the brain.9
Here, we present the clinical and immunological findings of two patients with PNP deficiency who were referred to the Immunology, Asthma and Allergy Research Institute (IAARI). They presented novel features of the disease and the result of this study may provide valuable evidence for early diagnosis.
Case Report
The first patient was a 20-month-old male and the third child of non-consanguineous parents referred to IAARI with recurrent respiratory tract infections, chronic diarrhea, rectal prolapse, and gastroesophageal reflux since infancy. He had a five-time history of hospital admissions since he was 15 months old for gastroenteritis, sepsis and severe anemia. The patient was regularly immunized with all standard vaccines including BCG and OPV-1 as live vaccinations without any complications.
He had developmental delay and his brain CT scan revealed mild cortical atrophy at one year of age. Diminished granulopoiesis in different myeloid precursors with mature granulocytes and a reduced number of erythroid precursors were shown in his bone marrow aspirate (BMA). Laboratory investigations showed Coombs-positive hemolytic anemia treated with several times blood transfusions. His immunological workup showed eosinophilia, lymphopenia, and low T cell numbers and function (Table 1). Biochemical evaluation revealed undetectable uric acid in his serum sample (Table 1). Based on his severe T cell lymphopenia, low uric acid and clinical findings, PNP deficiency was the most probable diagnosis. The genomic DNA of the patient was investigated for PNP gene mutation by PCR (using specific primers and an Amplicon PCR kit) and the product was subjected to Sanger sequencing (ABI 3730XL genetic analyzer, Applied Biosystems). The sequencing results were analyzed by the FinchTV program. A novel large homozygous deletion (c.246_285 + 9del, p. Gly83_Lys95 + 9 bp intron) was found in exon 3 of the PNP gene (NM_000270.4) (Figure 1A). This variant has been indicated as likely pathogenic based on VarSome tools.
Table 1.
Immunological and Biomedical Evaluations of two Patients with PNP Deficiency
|
Lab tests
|
Patient 1
values at 2 years (Normal range)a
|
Patient 2
values at 6 years (Normal range)a
|
| Hb (g/dL) |
8.8 (12.0 (10.5)) |
10.7 (12.5 (11.5)) |
|
WBC (103/µL)
|
5.49 (10.6 (6-17)) |
2.94 (8.5 (5-15.5)) |
|
NEUT (103/µL)
|
3.96 (1.5-8.5) |
1.29 (1.5-8) |
|
LYMPH (103/µL)
|
0.34 (3-9.5) |
0.26 (1.5-7) |
|
MONO (103/µL)
|
0.98 (0.5 mean) |
0.41 (0.4 mean) |
|
EOS (103/µL)
|
0.42 (0.04-0.4) |
1 (0.01-0.2) |
| CD3 count (%) |
14 (53-73) |
42.2 (60-79) |
| CD4 count (%) |
13 (32-51) |
25.7 (31-47) |
| CD8 count (%) |
9 (14-39) |
12.9 (18-35) |
| CD 19 count (%) |
14 (16-35) |
7.74 (16-35) |
| CD 16/56 count (n)* |
0.071 (0.18-0.92) |
0.14 (0.10-0.48) |
| IgE (Iu/mL) |
0.6 (0.31-29.5) |
160 (1.02-161.3) |
| IgM (mg/dL) |
223 (48-168) |
87 (48-207) |
| IgG (mg/dL) |
1863 (428-1051) |
1289 (633-1280) |
| IgA (mg/dL) |
63 (14-123) |
132 (33-202) |
| Isohemagglutinin titers |
1/32 (More than 1/8) |
1/2 (More than 1/8) |
| DTH to PPD antigen |
Negative |
- |
| DTH to DT antigen |
Negative |
- |
| TREC (copies/3.2 mmDBS) |
0 |
- |
| Uric acid (mg/dl) |
undetectable (2.4-6.8) |
1 (2.4-5.9) |
a Age-Related Reference Range from The Harriet Lane Handbook, Jason W. Custer and Rachel E. Rau, Eighteenth Edition.
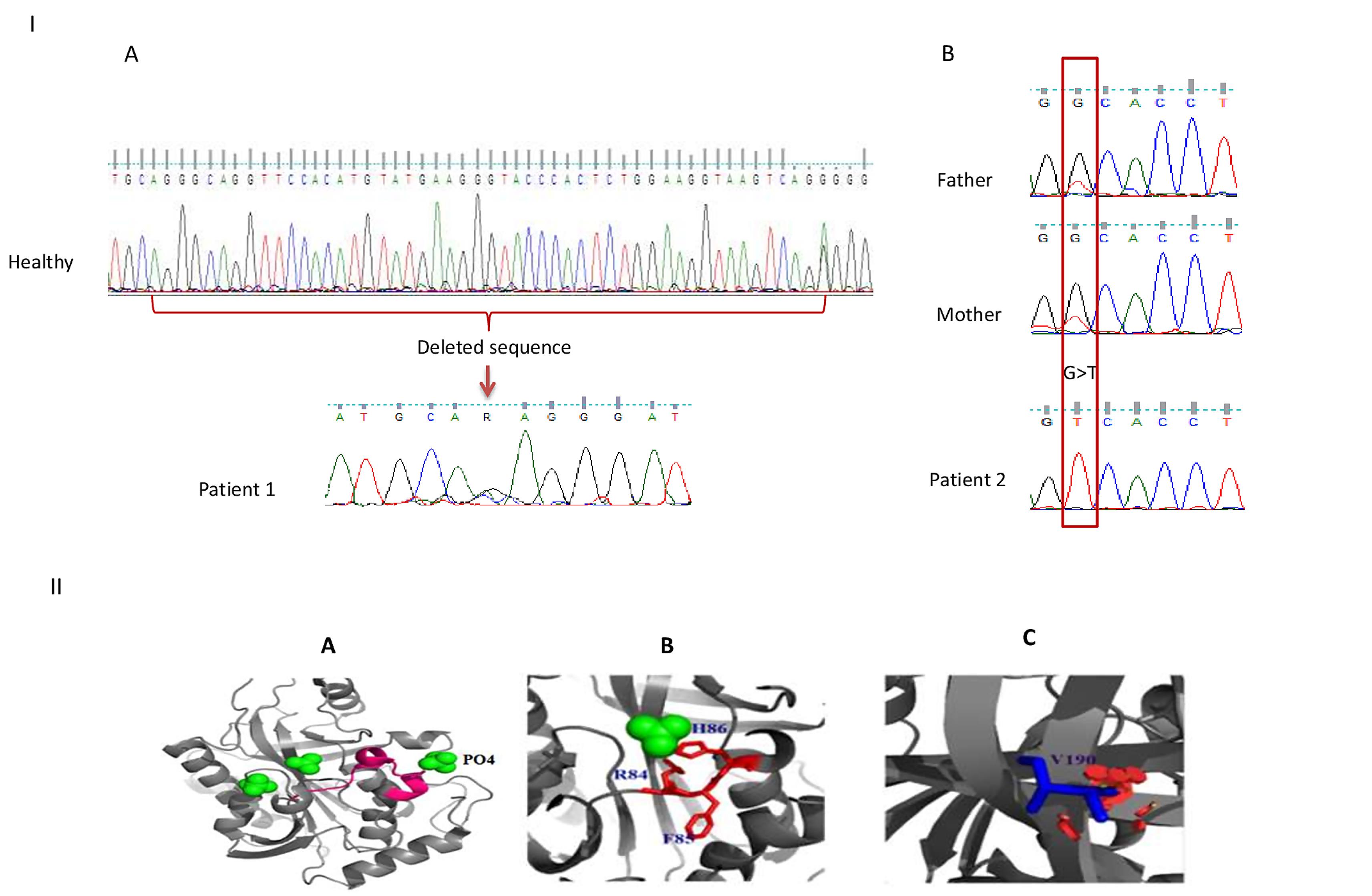
Figure 1.
PNP Gene Mutations and Ribbon Diagram of PNP Monomer. I) Schematic presentation of PNP gene mutations in patients. Deleted sequences (c.246_285 + 9del) in patient 1 compared to healthy control (A). Homozygote c.569G > T mutation in patient 2 and heterozygote mutation in his parents (B). II) Ribbon diagram for the monomer of PNP by PYMOL. The crystal structure at 2.3 Å resolution; PDB 1M73 was used as the model structure for the analysis. Structure visualization and mutagenesis analysis were performed with the PyMOL program (DeLano Scientific, San Carlos, CA, USA). Deletion of p. Gly83_Lys95 in patient 2 is shown in pink (A). The phosphate and sulfate binding site is deleted in patient 1 (B). Substitution of Gly to Val in patient 2 (C)
.
PNP Gene Mutations and Ribbon Diagram of PNP Monomer. I) Schematic presentation of PNP gene mutations in patients. Deleted sequences (c.246_285 + 9del) in patient 1 compared to healthy control (A). Homozygote c.569G > T mutation in patient 2 and heterozygote mutation in his parents (B). II) Ribbon diagram for the monomer of PNP by PYMOL. The crystal structure at 2.3 Å resolution; PDB 1M73 was used as the model structure for the analysis. Structure visualization and mutagenesis analysis were performed with the PyMOL program (DeLano Scientific, San Carlos, CA, USA). Deletion of p. Gly83_Lys95 in patient 2 is shown in pink (A). The phosphate and sulfate binding site is deleted in patient 1 (B). Substitution of Gly to Val in patient 2 (C)
The patient died because of a respiratory infection at 20 months of age. Unfortunately, the samples of the patient’s parents were not available.
The second patient was a six-year-old boy from consanguineous parents referred to IAARI due to recurrent infections. His physical examination showed that he suffered from general muscular hypotonia, disability to walk, and developmental delay from his infancy. He had a four-time history of hospital admissions because of sinusitis, pneumonia, fever, and diarrhea. At the age of 2.5 years, the patient developed Coombs-positive hemolytic anemia that was treated with blood transfusion, IVIG, and prednisolone. At the age of 6 years, his laboratory investigations revealed neutropenia. Leukopenia and lymphopenia were also noted in his BMA sample. Moreover, patient 2 had a history of eczema and cradle cap. The result of the allergic macroarray test revealed food allergy to eggs, hazelnut, pistachio nut, celery, and flour. The other immunological assessments showed normal serum immunoglobulin (Ig) levels, although the IgM isohemagglutinin titer did not show an efficient response (Table 1); therefore he had received IVIG regularly. The undetectable uric acid level in his serum sample suggested PNP deficiency. Next generation sequencing (NGS) of the whole exome was performed using SureSelect V6 Exome Kit (Illumina, San Diego, CA, USA) and the result showed a novel likely pathogenic missense mutation in exon 5 (c.569G > T) of the PNP gene (NM_000270.4) which changes Gly 190 to Val (Figure 1B). This variant has been mentioned as of uncertain significance based on VarSome (Pathogenic computational verdict based on 12 pathogenic predictions from BayesDel_addAF, DANN, DEOGEN2, EIGEN, FATHMM-MKL and 7 more vs 1 benign prediction from PrimateAI). The heterozygosity of this mutation in the patient’s parents was confirmed by Sanger sequencing. Moreover, the result of the TREC assay of the patient showed undetectable levels of TREC, confirming T cell deficiency. He received hematopoietic stem cell transplantation (HSCT) from his grandmother for the treatment and after 6 months, he is in good condition with complete engraftment. Moreover, his allergic reaction has significantly diminished clinically.
Discussion
Here, we introduce two Iranian patients in individual families with PNP deficiency and no family history of recurrent infections and immunologic disorders. Genetic analyses of these patients revealed a novel deletion in exon 3 and a novel missense mutation in Exon 5 in their PNP genes, respectively. The most common manifestation in PNP patients is recurrent infections, mainly respiratory tract infections. Defective mitochondrial function or depletion of dGTP in neurons result in neurologic abnormalities, hypotonia, tremors, retarded motor development, ataxia, and behavioral difficulties. Varying degrees of mental retardation have been reported in more than half of the PNP-deficient patients.2,8,10 Both patients in this study presented neurological defects, as well.
The defective transformation of inosine into hypoxanthine is suggestive of PNP deficiency which leads to hypouricemia. PNP deficiency could also be characterized by lymphopenia associated with low levels of uric acid and increased levels of inosine.7,11 Patient 1 manifested T cell deficiency, gastroenteritis, and pneumonia from early infancy.
In addition to the above-mentioned symptoms, patient 2 suffered from neutropenia, anemia, eosinophilia, and severe food allergy. Versatile anomalies and symptoms in these patients are related to the vital biological function of the PNP enzyme and purine metabolites.2 Fewer than half of PNP deficiency patients develop autoimmune diseases, notably systemic lupus erythematosus, idiopathic thrombocytopenic purpura and autoimmune hemolytic anemia, while both patients in this study suffered from anemia.12,13 It has been suggested that PNP deficiency results in increased activation of TLR7 and subsequently IL-6 up-regulation leading to autoimmunity.14
Previously, Dror et alreported a patient with PNP deficiency and eosinophilia.15 It is assumed that primary immunodeficiency (PID) could be a potential cause of eosinophilia, several primary immunodeficiencies including T cells deficiency, phagocytic dysfunction, and cytokine signaling disorders are reported to be related to eosinophilia; therefore, clinical evaluation of patients with eosinophilia has a considerable impact on early diagnosis of patients who may have immunodeficiencies.16,17
Recently, TREC/KREC assay has been proven as a tool for newborn screening to identify patients with primary immunodeficiency diseases including SCID and X-linked agammaglobulinemia (XLA).18 Therefore, early diagnosis of this disorder through TREC/KREC assay could be useful in the effective treatment of patients by allogeneic HSCT, enzyme replacement, and gene therapy.19,20 The result of the TREC assay of patient 2 showed undetectable levels of TREC, which follows the immunophenotyping findings of this patient. This is in agreement with the previous studies which showed that PNP deficiency could be detected by TREC assay.21,22
Previously, a missense PNP gene variant has been reported with no obvious effect on PNP activity and also normal uric acid level was found in an affected child with a mutation in the PNP gene.23 Moreover, developmental delay has been reported in two Iranian patients with PNP.24 However, a recent study showed no neurological defects and developmental delay in an Iranian PNP-deficient patient along with IgA deficiency.25 Therefore, a definite diagnosis of the disease is based on genetic analysis and measuring PNP activity. The investigated mutations in the current study are likely pathogenic based on the American College of Medical Genetics and Genomics (ACMG) guideline.26
PNP is a homotrimer protein and each monomer consist of 295 amino acids. The purine binding site is composed of Ala116, Phe200, Glu201, Val217, Met219, Thr242, and Asn243 residues.27 Besides, each monomer has 4 regulatory phosphate binding sites and 3 sulfate ligand binding sites. Patient 1 in this study had a large deletion Gly83_Lys95 + 9 bp intron in exon 3 which includes Arg84, Phe85, and His86 that comprise a phosphate binding site; Arg84 and His86 are also involved in sulfate binding site.28 This deletion extended through the splice site and aberrant splicing that leads to intron retention or exon skipping (Figure 1A, B). The second mutation in patient 2 changes the conserved glycine190 (an amino acid without a side chain) to valine which may change protein conformation and function, as well (Figure 1C).
It is notable to mention that no allele frequencies were reported in the 1000 genome project for these variations. We must acknowledge the limitations of our study when discussing the enzyme activity of PNP and the expression of its protein.
Conclusion
Due to the poor prognosis of PNP deficiency, early diagnosis and proper treatment are vital for these patients. Currently, HSCT is the only available cure; hopefully, other efficient treatments, notably gene therapy or enzyme replacement, will be developed for PNP-deficient patients.
Acknowledgements
The funding for this research was provided by the Immunology, Asthma and Allergy Research Institute, Tehran University of Medical Sciences.
Competing Interests
The authors declared that they have no conflict of interest.
Ethical Approval
The study was approved by the ethics committee of IAARI (#IR.TUMS.IAARI.REC.1399.004) and the written informed consent was taken from patients’ parents.
Funding
This study was supported by Immunology, Asthma & Allergy Research Institute and Tehran University of Medical Sciences.
References
- Grunebaum E, Cohen A, Roifman CM. Recent advances in understanding and managing adenosine deaminase and purine nucleoside phosphorylase deficiencies. Curr Opin Allergy Clin Immunol 2013; 13(6):630-8. doi: 10.1097/aci.0000000000000006 [Crossref] [ Google Scholar]
- Grunebaum E, Campbell N, Leon-Ponte M, Xu X, Chapdelaine H. Partial purine nucleoside phosphorylase deficiency helps determine minimal activity required for immune and neurological development. Front Immunol 2020; 11:1257. doi: 10.3389/fimmu.2020.01257 [Crossref] [ Google Scholar]
- Torun B, Bilgin A, Orhan D, Gocmen R, Kılıc SS, Kuskonmaz B. Combined immunodeficiency due to purine nucleoside phosphorylase deficiency: outcome of three patients. Eur J Med Genet 2022; 65(3):104428. doi: 10.1016/j.ejmg.2022.104428 [Crossref] [ Google Scholar]
- Markert ML. Purine nucleoside phosphorylase deficiency. Immunodefic Rev 1991; 3(1):45-81. [ Google Scholar]
- Aytekin C, Yuksek M, Dogu F, Yagmurlu A, Yildiran A, Fitoz S. An unconditioned bone marrow transplantation in a child with purine nucleoside phosphorylase deficiency and its unique complication. Pediatr Transplant 2008; 12(4):479-82. doi: 10.1111/j.1399-3046.2007.00890.x [Crossref] [ Google Scholar]
- Carson DA, Lakow E, Wasson DB, Kamatani N. Lymphocyte dysfunction caused by deficiencies in purine metabolism. Immunol Today 1981; 2(12):234-8. doi: 10.1016/0167-5699(81)90010-4 [Crossref] [ Google Scholar]
- Parvaneh N, Ashrafi MR, Yeganeh M, Pouladi N, Sayarifar F, Parvaneh L. Progressive multifocal leukoencephalopathy in purine nucleoside phosphorylase deficiency. Brain Dev 2007; 29(2):124-6. doi: 10.1016/j.braindev.2006.07.008 [Crossref] [ Google Scholar]
- Aytekin C, Dogu F, Tanir G, Guloglu D, Santisteban I, Hershfield MS. Purine nucleoside phosphorylase deficiency with fatal course in two sisters. Eur J Pediatr 2010; 169(3):311-4. doi: 10.1007/s00431-009-1029-6 [Crossref] [ Google Scholar]
- Schejter YD, Even-Or E, Shadur B, NaserEddin A, Stepensky P, Zaidman I. The broad clinical spectrum and transplant results of PNP deficiency. J Clin Immunol 2020; 40(1):123-30. doi: 10.1007/s10875-019-00698-1 [Crossref] [ Google Scholar]
- Madkaikar MR, Kulkarni S, Utage P, Fairbanks L, Ghosh K, Marinaki A. Purine nucleoside phosphorylase deficiency with a novel PNP gene mutation: a first case report from India. BMJ Case Rep 2011; 2011:bcr0920114804. doi: 10.1136/bcr.09.2011.4804 [Crossref] [ Google Scholar]
- Kütükçüler N, Bölük E, Tökmeci N, Karaca NE, Azarsız E, Aksu G. Recurrent infections, neurologic signs, low serum uric acid levels, and lymphopenia in childhood: purine nucleoside phosphorylase deficiency, an emergency for infants. Turk Pediatri Ars 2020; 55(3):320-7. doi: 10.14744/TurkPediatriArs.2019.83788 [Crossref] [ Google Scholar]
- Al-Saud B, Al Alawi Z, Hussain FB, Hershfield M, Alkuraya FS, Al-Mayouf SM. A case with purine nucleoside phosphorylase deficiency suffering from late-onset systemic lupus erythematosus and lymphoma. J Clin Immunol 2020; 40(6):833-9. doi: 10.1007/s10875-020-00800-y [Crossref] [ Google Scholar]
- Arduini A, Marasco E, Marucci G, Pardeo M, Insalaco A, Caiello I. An unusual presentation of purine nucleoside phosphorylase deficiency mimicking systemic juvenile idiopathic arthritis complicated by macrophage activation syndrome. Pediatr Rheumatol Online J 2019; 17(1):25. doi: 10.1186/s12969-019-0328-3 [Crossref] [ Google Scholar]
- Fox DA. Immunodeficiency and autoimmunity: companions not opposites. J Clin Invest 2022; 132(16):e162170. doi: 10.1172/jci162170 [Crossref] [ Google Scholar]
- Dror Y, Grunebaum E, Hitzler J, Narendran A, Ye C, Tellier R. Purine nucleoside phosphorylase deficiency associated with a dysplastic marrow morphology. Pediatr Res 2004; 55(3):472-7. doi: 10.1203/01.pdr.0000111286.23110.f8 [Crossref] [ Google Scholar]
- Navabi B, Upton JE. Primary immunodeficiencies associated with eosinophilia. Allergy Asthma Clin Immunol 2016; 12:27. doi: 10.1186/s13223-016-0130-4 [Crossref] [ Google Scholar]
- Williams KW, Milner JD, Freeman AF. Eosinophilia associated with disorders of immune deficiency or immune dysregulation. Immunol Allergy Clin North Am 2015; 35(3):523-44. doi: 10.1016/j.iac.2015.05.004 [Crossref] [ Google Scholar]
- Borte S, von Döbeln U, Fasth A, Wang N, Janzi M, Winiarski J. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood 2012; 119(11):2552-5. doi: 10.1182/blood-2011-08-371021 [Crossref] [ Google Scholar]
- Puck JM. Population-based newborn screening for severe combined immunodeficiency: steps toward implementation. J Allergy Clin Immunol 2007; 120(4):760-8. doi: 10.1016/j.jaci.2007.08.043 [Crossref] [ Google Scholar]
- Alizadeh Z, Dashti P, Mazinani M, Nourizadeh M, Shakerian L, Tajik S. Clinical and genetic study of X-linked agammaglobulinemia patients (the benefit of early diagnosis). Iran J Allergy Asthma Immunol 2020; 19(3):305-9. doi: 10.18502/ijaai.v19i3.3458 [Crossref] [ Google Scholar]
- van der Spek J, Groenwold RH, van der Burg M, van Montfrans JM. TREC based newborn screening for severe combined immunodeficiency disease: a systematic review. J Clin Immunol 2015; 35(4):416-30. doi: 10.1007/s10875-015-0152-6 [Crossref] [ Google Scholar]
- Martín-Nalda A, Rivière JG, Català-Besa M, García-Prat M, Parra-Martínez A, Martínez-Gallo M. Early diagnosis and treatment of purine nucleoside phosphorylase (PNP) deficiency through TREC-based newborn screening. Int J Neonatal Screen 2021; 7(4):62. doi: 10.3390/ijns7040062 [Crossref] [ Google Scholar]
- Al-Saud B, Alsmadi O, Al-Muhsen S, Al-Ghonaium A, Al-Dhekri H, Arnaout R. A novel mutation in purine nucleoside phosphorylase in a child with normal uric acid levels. Clin Biochem 2009; 42(16-17):1725-7. doi: 10.1016/j.clinbiochem.2009.08.017 [Crossref] [ Google Scholar]
- Parvaneh N, Teimourian S, Jacomelli G, Badalzadeh M, Bertelli M, Zakharova E. Novel mutations of NP in two patients with purine nucleoside phosphorylase deficiency. Clin Biochem 2008; 41(4-5):350-2. doi: 10.1016/j.clinbiochem.2007.11.007 [Crossref] [ Google Scholar]
- Fekrvand S, Yazdani R, Abolhassani H, Ghaffari J, Aghamohammadi A. The first purine nucleoside phosphorylase deficiency patient resembling IgA deficiency and a review of the literature. Immunol Invest 2019; 48(4):410-30. doi: 10.1080/08820139.2019.1570249 [Crossref] [ Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17(5):405-24. doi: 10.1038/gim.2015.30 [Crossref] [ Google Scholar]
- Canduri F, dos Santos DM, Silva RG, Mendes MA, Basso LA, Palma MS. Structures of human purine nucleoside phosphorylase complexed with inosine and ddI. Biochem Biophys Res Commun 2004; 313(4):907-14. doi: 10.1016/j.bbrc.2003.11.179 [Crossref] [ Google Scholar]
- de Azevedo WF Jr, Canduri F, dos Santos DM, Silva RG, de Oliveira JS, de Carvalho LP. Crystal structure of human purine nucleoside phosphorylase at 23A resolution. Biochem Biophys Res Commun 2003; 308(3):545-52. doi: 10.1016/s0006-291x(03)01431-1 [Crossref] [ Google Scholar]