Arch Iran Med. 26(6):346-354.
doi: 10.34172/aim.2023.52
Original Article
Somatic Mutation of PRKAR1A in Four Cases of Sporadic Cardiac Myxoma
Yunpeng Sun Conceptualization, Data curation, Formal analysis, Methodology, Resources, Writing – original draft, Writing – review & editing, 1, # 
Zhiping Li Conceptualization, Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing, 2, #
Jingnan Sun Data curation, Resources, Writing – review & editing, 3
Dashi Ma Data curation, Methodology, Writing – review & editing, 1
Xue Shan Data curation, Methodology, Writing – review & editing, 1
Xia Chen Conceptualization, Formal analysis, Investigation, Project administration, Supervision, Validation, Writing – review & editing, 2, *
Author information:
1Department of Cardiac Surgery, The First Hospital of Jilin University, Changchun, Jilin, 130021, China
2Department of Pharmacology, Basic Medical College of Jilin University, Changchun, Jilin, 130021, China
3Department of Hematology, The First Hospital of Jilin University, Changchun, Jilin, 130021, China
#Yunpeng Sun and Zhiping Li contributed equally as co-first authors.
Abstract
Background:
Inactivating mutations of the protein kinase A regulatory subunit 1 alpha (PRKAR1A) gene have been reported in familial cardiac myxoma. However, the role of PRKAR1A mutation in sporadic cardiac myxoma remains unknown.
Methods:
Targeted next-generation sequencing (NGS) was performed to identify mutations with the PRKAR1A gene in seven cases of sporadic cardiac myxomas. Sanger sequencing of DNA from cardiac myxoma specimens and matched peripheral blood samples was performed to verify the identified mutations.
Results:
Targeted NGS of myxoma DNA revealed 232 single nucleotide variants in 141 genes and 38 insertion-deletion mutations in 13 genes. Six PRKAR1A mutations were identified in four of the seven cardiac myxoma cases, and thus, the PRKAR1A inactivating mutation rate was 57.2% (4/7, 95% CI=0.44-0.58, P<0.05). The PRKAR1A variants identified by Sanger sequencing analysis were consistent with those from the NGS analysis for the four myxoma specimens. All of the pathogenic PRKAR1A mutations led to premature termination of PRKAR1A, except for one synonymous mutation. Moreover, none of the nonsense and missense mutations found in the myxoma specimens were found in the matched peripheral blood samples.
Conclusion:
Pathogenic mutations of the PRKAR1A gene were identified in tumor specimens from four cases of sporadic cardiac myxoma, and the absence of these mutations in peripheral blood samples demonstrated that they were somatic mutations.
Keywords: Cardiac myxoma, PRKAR1A gene, Somatic mutation, Sporadic
Copyright and License Information
© 2023 The Author(s).
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article as: Sun Y, Li Z, Sun J, Ma D, Shan X, Chen X. Somatic mutation of PRKAR1A in four cases of sporadic cardiac myxoma. Arch Iran Med. 2023;26(6):346-354. doi: 10.34172/aim.2023.52
Introduction
Cardiac myxoma is the most common primary cardiac neoplasm in adults.1 On autopsy, myxomas account for almost half of the cardiac tumors found in 0.17% of the population.2 In approximately 75% of the general patient population, this tumor is located in the left atrium and arises from the atrial septum by the stalk.3,4 Cardiac myxomas are more likely to occur between the ages of 40 and 60 years.5 The clinical symptoms and signs of myxoma include fever of unknown origin, obstruction, and embolization, and although most of them are benign, they can be life-threatening.6 To date, the pathogenesis of cardiac myxoma remains unclear.
The PRKAR1A (OMIM 188830) gene encodes the regulatory subunit type 1 alpha (Riα) of the cyclic AMP (cAMP)-dependent protein kinase A (PKA) gene and acts as a tumor suppressor gene.7,8 Inactivating mutations ofthe PRKAR1A gene are found in familial cases of cardiac myxoma.9 Carney complex (CNC) (OMIM 160980) is an inheritable and autosomal dominant condition caused by germline inactivating mutations of the PRKAR1A gene.10,11 Carney et al first defined CNC as “the syndrome of myxomas, spotty pigmentation, and endocrine overactivity” in 1985.12 A pathogenic variant in PRKAR1A can be identified and used for diagnosing CNC,13 given that inactivating mutations of PRKAR1A were shown to be responsible for more than 70% of cases among patients a with familial history of CNC.14 More than 100 different mutations throughout the coding region of PRKAR1A have been reported.15 However, only a relatively small proportion of cardiac myxoma cases are diagnosed as CNC with an established familial pattern of inheritance. Instead, the majority of cardiac myxomas (90%) occur sporadically.16 Whether a mutation in the PRKAR1A gene is the cause of sporadic cardiac myxomas in patients without a relevant family history and without CNC is still controversial.4,17,18 In this study, mutations of PRKAR1A were studied in seven cases of sporadic cardiac myxomas that occurred in the absence of a family history of cardiac myxomas and without CNC.
Materials and Methods
Study Participants
The protocol for this study was reviewed and approved by the Human Medical Ethical Review Committee of the local hospital (The First Hospital of Jilin University, Changchun, China). All the patients were recruited in the Department of Cardiac Surgery, The First Hospital of Jilin University between January 2015 and January 2017 and provided written informed consent for participation. At the onset, the seven patients with cardiac myxomas recruited in this study had no known family history of cardiac myxomas or CNC. Cardiac myxoma specimens and blood samples were collected in cryogenic vials and immediately placed in liquid nitrogen after excision before transfer to a –80°C refrigerator for storage.
Histology
The cardiac myxoma specimens were fixed in 4% paraformaldehyde for 24 hours, washed in phosphate-buffered solution (PBS), and then embedded in paraffin. Hematoxylin and eosin (H&E) staining was used for pathological diagnosis as described previously.19 Immunohistochemistry analysis of cardiac myxoma specimens was not performed.
Targeted Next-generation Sequencing (NGS) Analysis
Genomic DNA was isolated using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). DNA samples were quantified with a Qubit Fluorometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) before the preparation of a library for NGS. The targeted capture genes are listed in Table S1 (Supplementary file 1). Targeted NGS was performed using a Solexa HiSeq 2000 sequencer (Illumina, San Diego, CA, USA). The target genes of the Cancer Risk Cap kit with a hybridization biotin-probe (MyGenostics, Inc, Beijing, China) were hybridized to exon DNA, and formatting was achieved by biotin-streptavidin capture of exon DNA. The sequencing depth was 340 times, and the coverage of the targeted genes was 98.8%. The low-quality reads and 30 and 50 adapters were filtered using Trim Galore (V0.3.5). The filtered reads with a sequencing quality > 20 and read length > 80 were aligned to the reference genome using the Burrows-Wheeler Aligner (V0.7.7).20 Duplicate reads were removed using Sequence Alignment/Map tools (V1.1).15,21 Single nucleotide variants (SNVs) were identified using UnifiedGenotyper of the GATK (V3.0) program with the dbSNP archive (version 138), using hg19 as the reference genome. Insertion-deletions (INDELs) were identified using the IndelRealigner of GATK (V3.0) and filtered through a 1000-genome database (http://www.1000genomes.org). Genetic variants were annotated across the genome using the ANNOVAR software (V2013Aug23) with respect to mutation type (missense, nonsense, or frameshift) and location (exonic, intronic, or intergenic region).15,21 The pathogenicity of the identified variants was assessed according to the “Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer” published in 2017.22
Sanger Sequencing Verification
Sanger sequencing was performed to verify the results of the targeted NGS analysis. Cardiac myxoma samples and matched peripheral blood samples from all seven cases were used for exon capture sequencing. Amplicon sequencing was carried out by Sanger sequencing. The polymerase chain reaction (PCR) primers were designed to produce a product of about 200 bp with the mutation upstream and downstream of each PRKAR1A gene. The sequencing primers used are listed in Table S2 (Supplementary file 1).
Statistical Analysis
The demographic and clinical data are presented as mean ± standard deviation (SD). Patient age, tumor diameter, tumor volume, and genotype frequencies were recorded or calculated, and 95% confidence intervals (CIs) were calculated using SPSS 25.0. Each experiment was repeated at least three times. The Mann-Whitney U test was used for comparisons between two samples. A difference was considered statistically significant with P < 0.05.
Results
Clinical Data
The clinical characteristics of the seven patients with sporadic cardiac myxoma are described in Table 1. Two myxoma patients were male, and five were female. The average age of the patients was 52.85 ± 10.2 years (95% CI = 0.47-0.61 years). The mean age of the women was significantly lower than that of the men (49.0 ± 9.49 years vs 62.5 ± 2.12 years, P < 0.05). All seven cardiac tumor specimens exhibited the typical pathological changes of myxoma and were diagnosed as cardiac myxomas (Figure 1). All the myxomas were located on the left side of the atrium. The average tumor diameter was 44 ± 12.47 mm (range, 23–60 mm; 95% CI = 0.32-0.56 mm). Most of the cardiac myxomas were nearly round or oval, and thus, tumor volume was calculated based on a spherical shape. Accordingly, the average tumor volume was 73.26 ± 45.56 cm3 (range, 6.37–113.04 cm3; 95% CI = 0.41-1.02 cm3).
Table 1.
Clinical Characteristics of Seven Cases with Sporadic Cardiac Myxomas
|
No.
|
Gender
|
Age (y)
|
Diameter (mm)
|
Volume (cm3)
|
Position
|
| Case 1 |
Male |
64 |
40 |
33.49 |
Atrial septum bottom |
| Case 2 |
Female |
34 |
50 |
65.62 |
Atrial septum |
| Case 3 |
Male |
61 |
60 |
113.04 |
Atrial septum |
| Case 4 |
Female |
56 |
54 |
82.41 |
Atrial septum |
| Case 5 |
Female |
58 |
23 |
6.37 |
Atrial septum top |
| Case 6 |
Female |
47 |
35 |
22.44 |
Atrial septum |
| Case 7 |
Female |
50 |
46 |
50.94 |
Atrial septum bottom |
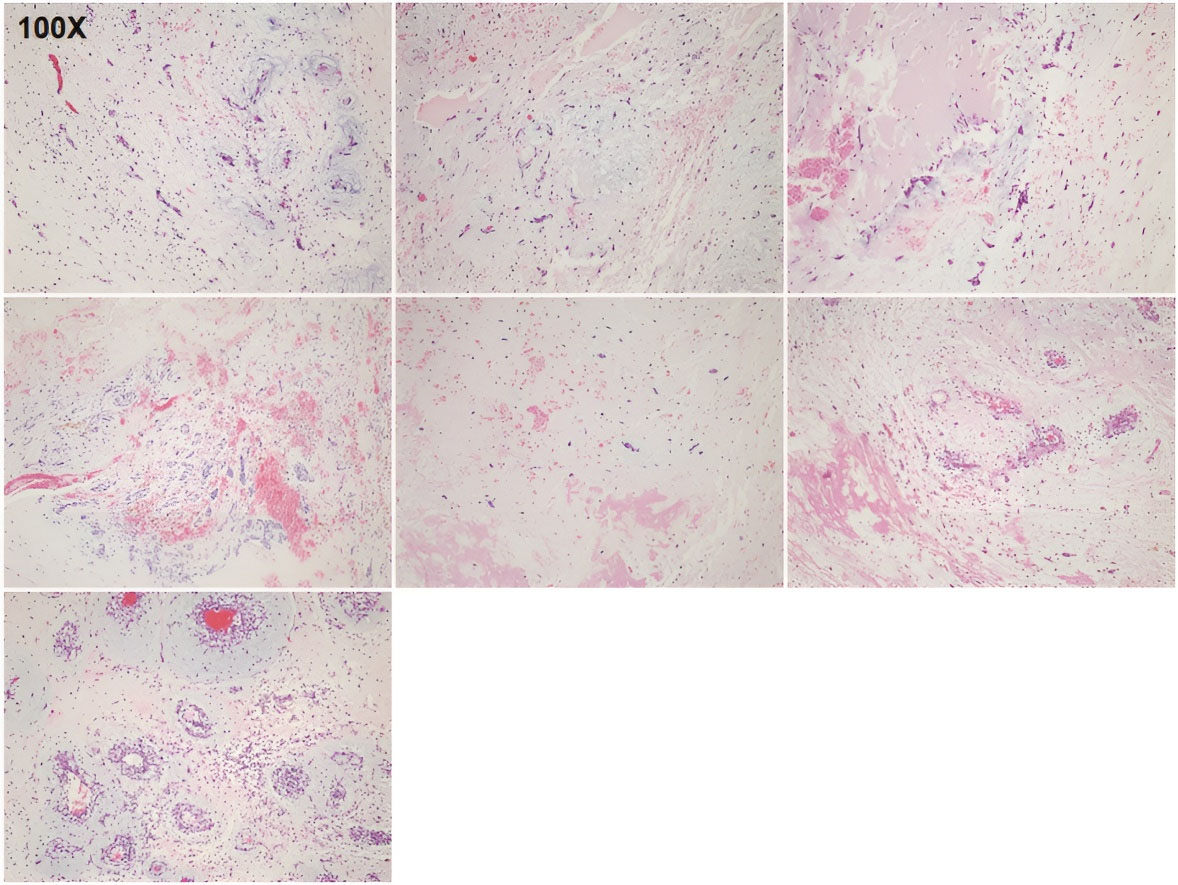
Figure 1.
Pathology of Seven Sporadic Cardiac Myxoma Specimens. Hematoxylin and eosin (H&E) staining was performed on sporadic cardiac myxoma samples from seven cases (Case 1 to Case 7). Magnification, 100 ×
.
Pathology of Seven Sporadic Cardiac Myxoma Specimens. Hematoxylin and eosin (H&E) staining was performed on sporadic cardiac myxoma samples from seven cases (Case 1 to Case 7). Magnification, 100 ×
Targeted NGS Analysis of Seven Sporadic Cardiac Myxomas
Using DNA extracted from the seven cardiac myxoma specimens, targeted NGS analysis identified 232 SNVs in 141 genes, which included 87 synonymous mutations, 132 missense mutations, 3 nonsense mutations, and 10 splicing mutations. INDEL variant analysis then demonstrated that 38 gene mutations (3 frameshift mutations and 35 non-frameshift mutations) were distributed in 13 genes (Table S3).
From the results of this targeted NGS analysis, we highlighted all the genes with a significant number of events. We identified RAI1 gene mutations in all seven cases, including two synonymous mutations RAI 1:c.840G > A (p.Q280Q) and RAI1:c.3885G > T(p.P1295P) and five non-frameshift mutations RAI1:c.832_834del(p.278_278del), RAI1:c.835_837del(p.279_279del), RAI1:c.832_837del (p.278_279del), RAI1:c.832_843del (p.278_281del), and RAI1:c.834_835insCAA (p.Q278delinsQQ). Additionally, eight mutations of the AR gene were found in five cases, and all of them led to non-frameshift mutations AR:c.170_171insGCA (p.L57delinsLQ), AR:c.171_173del (p.57_58del), AR:c.173_174insGCAGCA(p.Q58delinsQQQ), AR:c.1369_1371del (p.457_457del), AR:c.1369_1377del (p.457_459del), AR:c.1377_1378insGGC (p.G459delinsGG), AR:c.171_179del (p.57_60del), and AR:c.170_171insGCAGCA (p.L57delinsLQQ). Therefore, we calculated that the mutation rate of the AR gene was 71.4% (5/7; 95% CI = 0.39-0.78). Furthermore, we obtained similar results for non-frameshift mutations from the TBP INDEL variants TBP:c.163_171del (p.55_57del)and TBP:c.223_231 (p.75_77del) in six cases, and the mutation rate was 85.7% (6/7; 95% CI = 0.50-1.00, P < 0.05). The AKAP9 gene mutation involved three missense mutations AKAP 9:c.11135G > A (p.R3712Q), AKAP9:c.3827G > A (p.R1276Q), and AKAP9: c.5725G > A (p.A1909T) and one synonymous mutation AKAP9: c.10845G > A (p.K3615K). We identified AKAP9 gene mutations in four cases, for an AKAP9 mutation rate of 57.2% (4/7; 95% CI = 0.44-0.58, P < 0.05). Five mutations of the GIGYF2 gene were found among four cases, including one missense mutation GIGYF 2:c.3445C > A (p.P1149T), two non-frameshift mutations GIGYF2:c.3629_3630insGCA (p.P1210delinsPQ) and GIGYF2:c.3611_3612insGCA (p.P1204delinsPQ), and two synonymous mutations GIGYF2:c.297T > C (p.A99A)and GIGYF2:c.3612A > G (p.P1204P). Therefore, the mutation rate of GIGYF2 was also 57.2% (4/7; 95% CI = 0.44-0.58, P < 0.05). Still three other genes (DCTN1, GALC and GALNS) were mutated in two cases, and the mutation types were missense mutation, synonymous mutation, and splicing mutation (Table 2).
Table 2.
Next-generation Sequencing of Multiple Genes in Sporadic Cardiac Myxoma Specimens.
|
Gene
|
No.
|
Mutation
|
Amino acid alteration
|
Mutation type
|
|
RAI1
|
Case 1 |
c.840G > A |
p.Q280Q |
Synonymous |
| c.3885G > T |
p.P1295P |
Synonymous |
| c.832_834del |
p.278_278del |
Non-frameshift |
| Case 2 |
c.835_837del |
p.279_279del |
Non-frameshift |
| Case 3 |
c.840G > A |
p.Q280Q |
Synonymous |
| c.832_837del |
p.278_279del |
Non-frameshift |
| Case 4 |
c.840G > A |
p.Q280Q |
Synonymous |
| c.832_837del |
p.278_279del |
Non-frameshift |
| Case 5 |
c.832_843 |
p.278_281del |
Non-frameshift |
| Case 6 |
c.840G > A |
p.Q280Q |
Synonymous |
| c.832_834del |
p.278_278del |
Non-frameshift |
| Case 7 |
c.840G > A |
p.Q280Q |
Synonymous |
| c.832_834del |
p.278_278del |
Non-frameshift |
| c.834_835insCAA |
p.Q278delinsQQ |
Non-frameshift |
|
AR
|
Case 1 |
c.170_171insGCA |
p.L57delinsLQ |
Non-frameshift |
| Case 2 |
c.171_173del |
p.57_58del |
Non-frameshift |
| c.173_174insGCAGCA |
p.Q58delinsQQQ |
Non-frameshift |
| c.1369_1371del |
p.457_457del |
Non-frameshift |
| Case 3 |
c.171_173del |
p.57_58del |
Non-frameshift |
| c.1369_1377del |
p.457_459del |
Non-frameshift |
| c.1377_1378insGGC |
p.G459delinsGG |
Non-frameshift |
| Case 4 |
c.171_179del |
p.57_60del |
Non-frameshift |
| c.1369_1371del |
p.457_457del |
Non-frameshift |
| Case 6 |
c.170_171insGCAGCA |
p.L57delinsLQQ |
Non-frameshift |
| c.1369_1377del |
p.457_459del |
Non-frameshift |
|
AKAP9
|
Case 1 |
c.11135G > A |
p.R3712Q |
Missense |
| Case 3 |
c.10845G > A |
p.K3615K |
Synonymous |
| Case 4 |
c.3827G > A |
p.R1276Q |
Missense |
| Case 6 |
c.5725G > A |
p.A1909T |
Missense |
|
GIGYF2
|
Case 2 |
c.3629_3630insGCA |
p.P1210delinsPQ |
Non-frameshift |
| Case 3 |
c.297T > C |
p.A99A |
Synonymous |
| c.3612A > G |
p.P1204P |
Synonymous |
| c.3611_3612insGCA |
p.P1204delinsPQ |
Non-frameshift |
| Case 4 |
c.3445C > A |
p.P1149T |
Missense |
| Case 6 |
c.3612A > G |
p.P1204P |
Synonymous |
|
|
c.3611_3612insGCA |
p.P1204delinsPQ |
Non-frameshift |
|
TBP
|
Case 1 |
c.163_171del |
p.55_57del |
Non-frameshift |
| Case 2 |
c.223_231del |
p.75_77del |
Non-frameshift |
| Case 3 |
c.163_171del |
p.55_57del |
Non-frameshift |
| Case 4 |
c.163_171del |
p.55_57del |
Non-frameshift |
| Case 6 |
c.163_171del |
p.55_57del |
Non-frameshift |
| Case 7 |
c.163_171del |
p.55_57del |
Non-frameshift |
|
DCTN1
|
Case 1 |
c.1811A > G |
p.Q604R |
Missense |
| Case 2 |
c.155C > T |
p.P52L |
Missense |
|
GALC
|
Case 1 |
c.645C > T |
p.L215L |
Synonymous |
| Case 3 |
c.1832T > C |
p.L611S |
Missense |
|
GALNS
|
Case 2 |
c.566 + 5T > C |
splicing |
Splicing |
| Case 4 |
c.566 + 5T > C |
splicing |
Splicing |
|
PRKAR1A
|
Case 2 |
c.61_62insAC |
p.Y21fs |
Frameshift |
| Case 3 |
c.273_274insAAAG |
p.V91fs |
Frameshift |
| Case 6 |
c.496C > T |
p.Q166X |
Missense |
|
|
c.569delG |
p.W190fs |
Frameshift |
| Case 7 |
c.366C > G |
p.Y122X |
Nonsense |
|
|
c.678C > T |
p.I226I |
Synonymous |
Finally, six PRKAR1A genemutations were found across four cases, including one missense mutation PRKAR1 A:c.496C > T (p.Q166X), one synonymous mutation PRKAR1A:c.678C > T (p.I226I), one nonsense mutation PRKAR1A:c.366C > G (p.Y122X), and three frameshiftmutations PRKAR1A:c.61_62insAC (p.Y21fs), PRKAR1A:c.273_274insAAAG (p.V91fs), and PRKAR1A:c.569delG (p.W190fs). The missense mutation, nonsense mutation, and frameshift mutation lead to premature appearance of terminations and eventually give rise to gene mutations (Table 2).
PRKAR1A Mutations in Cases of Sporadic Cardiac Myxoma
As described above, targeted NGS detection was performed for the seven cases of sporadic cardiac myxoma. From the analysis of SNVs and INDELs, the following six PRKAR1A mutations were detected across four cases: PRKAR1 A:c.61_62insAC (p.Y21fs), PRKAR1A:c.273_274insAAAG (p.V91fs), PRKAR1A:c.496C > T (p.Q166X), PRKAR1A:c.569delG (p.W190fs), PRKAR1A:c.366C > G (p.Y122X), and PRKAR1A:c.678C > T (p.I226I). The three frameshift mutations of PRKAR1 A:c.61_62insAC (p.Y21fs), PRKAR1A:c.273_274insAAAG (p.V91fs), and PRKAR1A:c.569delG (p.W190fs) occurred in the chromosome (Chr) 17 region 66511601 of Case 2, the Chr 17 region 66518992 of Case 3, and the Chr 17 region 66521914 of Case 6 separately. The PRKAR1 A:c.678C > T (p.I226I) and PRKAR1A:c.366C > G (p.Y122X) mutations were found in the myxoma specimen of Case 7 and were located in the Chr 17 region 66522023 and Chr 17 region 66519883. Notably, the two mutations in Case 7 had differing significance, as one was a synonymous mutation and the other a nonsense mutation. chr17:66520212 (PRKAR1A ):c.496C > T (p.Q166X)of Case 6 resulted in a missense mutation (Figure 2a). Because all three types of mutations (nonsense, missense and frameshift) lead to premature termination of PRKAR1A, these mutations found in these four cases were PRKAR1A pathogenic mutations (Figure 2b). Therefore, the PRKAR1A inactivating mutation rate among the seven cases with sporadic cardiac myxomas was 57.2% (4/7; 95% CI = 0.44-0.58). These findings demonstrate that PRKAR1A mutation is a pathogenic mechanism of non-familial, sporadic cardiac myxoma in the absence of CNC.
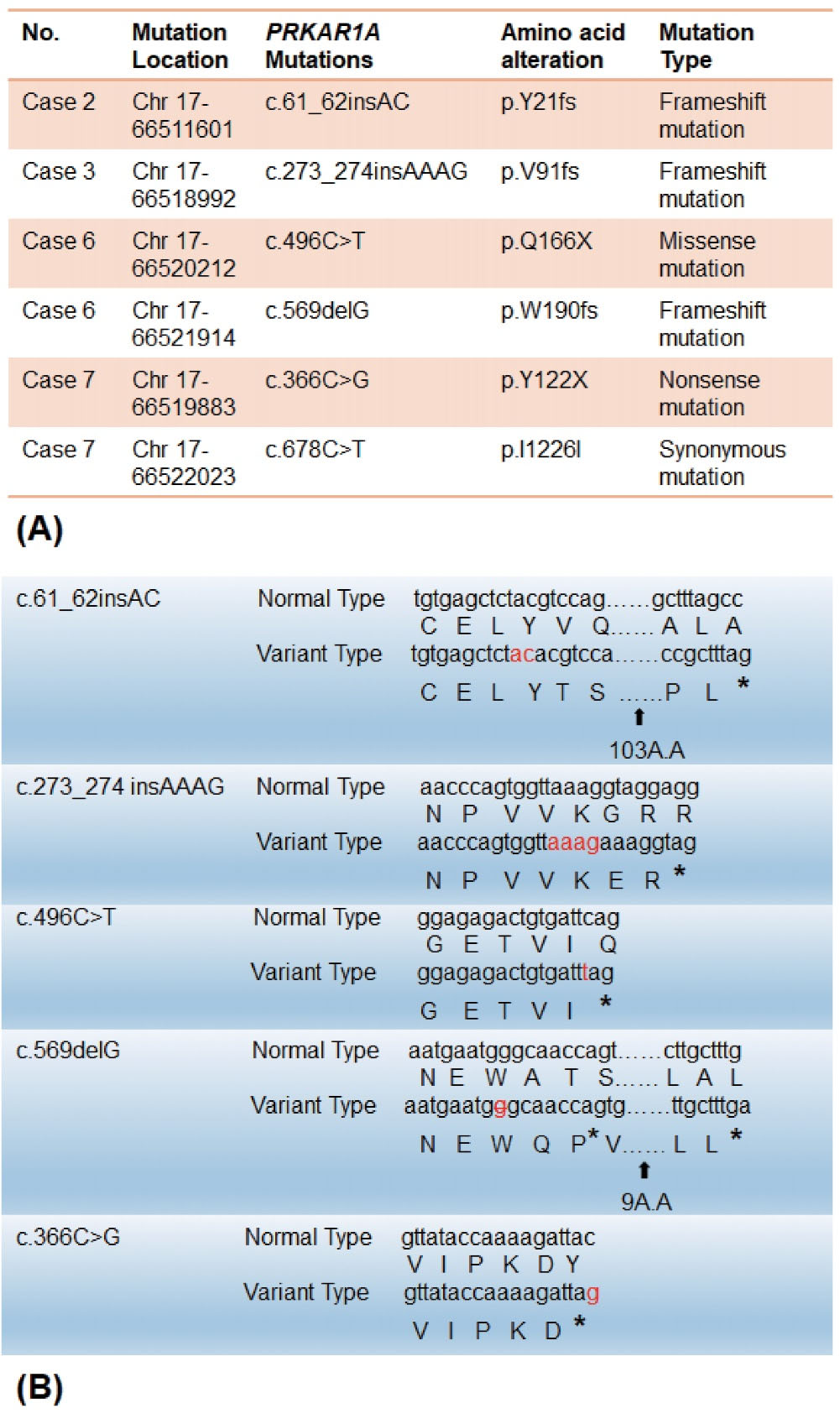
Figure 2.
Analysis of the Pathogenic PRKAR1A Mutations. (A) Targeted next-generation sequencing (NGS) identified six PRKAR1A mutations in four cases of sporadic cardiac myxoma. (B) Five pathogenic PRKAR1A mutations showed a difference between variant type and normal type, whereas one PRKAR1A mutation was a synonymous mutation. * indicates the terminators; chr: chromosome
.
Analysis of the Pathogenic PRKAR1A Mutations. (A) Targeted next-generation sequencing (NGS) identified six PRKAR1A mutations in four cases of sporadic cardiac myxoma. (B) Five pathogenic PRKAR1A mutations showed a difference between variant type and normal type, whereas one PRKAR1A mutation was a synonymous mutation. * indicates the terminators; chr: chromosome
Sanger Sequencing Verification in Myxoma Tissue and Matched Peripheral Blood
The six mutations of PRKAR1A detected in the four cases were verified by Sanger sequencing of both cardiac myxoma specimens and matched peripheral blood samples. Primers were designed to be located upstream and downstream of each PRKAR1A mutation (approximately 200 bp in length). Sanger sequencing analysis of myxoma samples identified six PRKAR1A mutations that were consistent with the PRKAR1A variant mutations detected by targeted NGS analysis (Figure 3). However, no nonsense, missense, or frameshift mutations were found in the matched peripheral blood samples from the four cases, with the exception of one synonymous mutation (c.678C > T) (Figure 3). Because pathogenic mutations in PRKAR1A were found in four of the seven (57.2%; 95% CI = 0.44-0.58, P < 0.05) cardiac myxoma samples and in none of the matched peripheral blood samples, the PRKAR1A mutations in these sporadic cardiac myxomas were considered somatic mutations, which were acquired and non-inheritable.
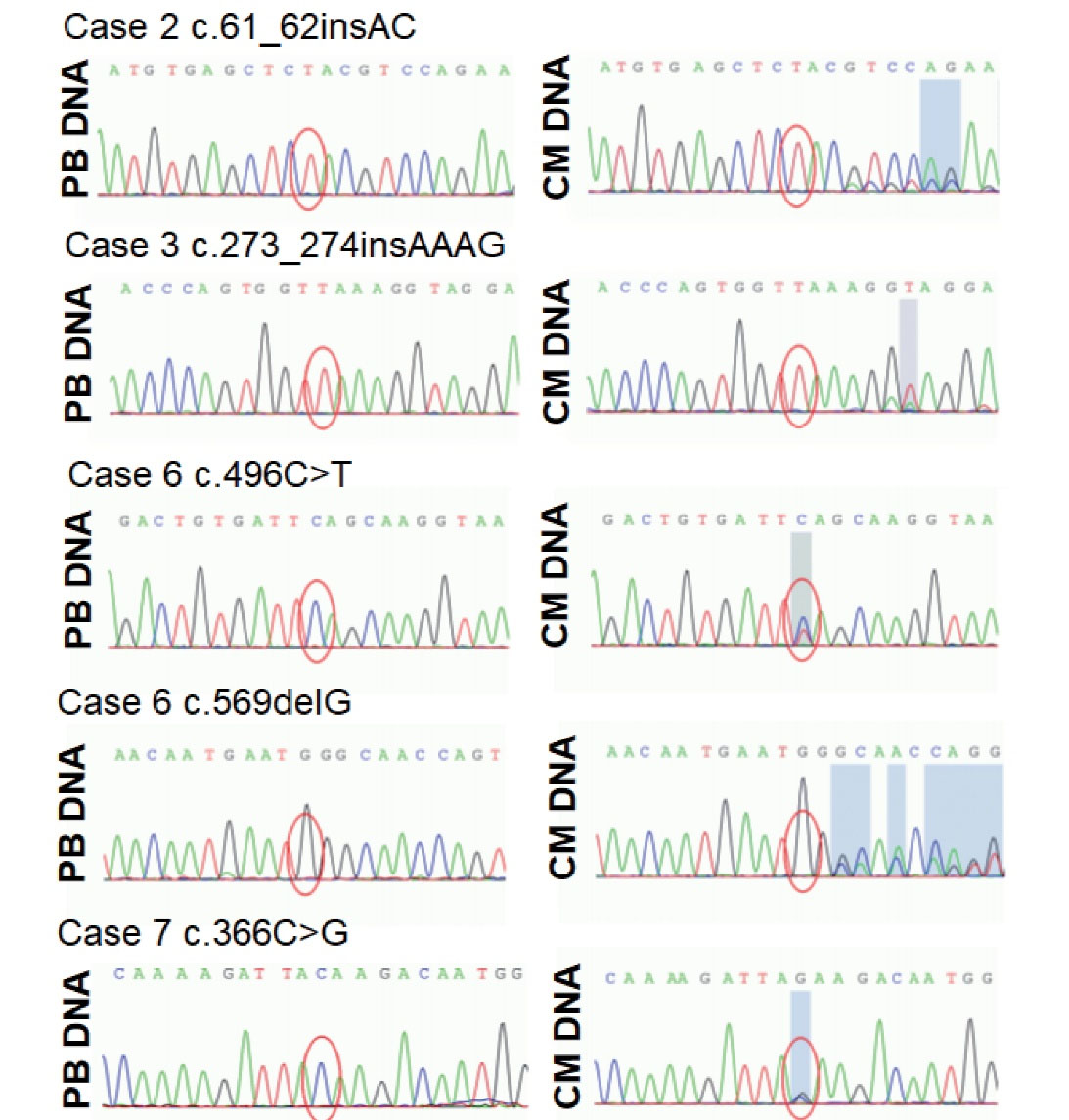
Figure 3.
Sanger Sequencing Analysis of Cardiac Myxoma Specimens and Matched Peripheral Blood Samples from the Four Cases with Sporadic Cardiac Myxoma with Pathogenic PRKAR1A Mutations Identified by Targeted NGS. Five pathogenic PRKAR1A mutations were found in the cardiac myxomas. None of the pathogenic mutations were found in the matched peripheral blood samples. CM, cardiac myxoma; PB, peripheral blood.
.
Sanger Sequencing Analysis of Cardiac Myxoma Specimens and Matched Peripheral Blood Samples from the Four Cases with Sporadic Cardiac Myxoma with Pathogenic PRKAR1A Mutations Identified by Targeted NGS. Five pathogenic PRKAR1A mutations were found in the cardiac myxomas. None of the pathogenic mutations were found in the matched peripheral blood samples. CM, cardiac myxoma; PB, peripheral blood.
Discussion
The World Health Organization defines cardiac myxoma as a common benign tumor composed of stromal cells scattered in the myxoid matrix.9 The incidence of primary cardiac tumors is reported to be 0.0017%–0.19%, approximately 75% of which are benign, and cardiac myxomas account for about 50% of primary benign tumors.23 Sporadic cardiac myxomas occur most commonly in middle-aged women, are generally single lesions on the left atrial aspect of the interatrial septum, and do not recur after surgical resection.24 In the present study, all seven cardiac myxomas were located on the left atrium, and none of the seven patients had a history of surgery for myxoma or CNC. Although the sample size was small, the number of women was greater than men, and the age of the women was lower than that of the men. The pathological diagnosis of myxoma for all seven sporadic tumors was confirmed by H&E staining (Figure 1).
DNA was extracted from the seven cardiac myxoma specimens for genetic analysis by targeted NGS. Mutations that were found at a high incidence in these seven cases were compared with the mutations in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (https://cancer.sanger.ac.uk/cosmic). We were surprised to find that the most frequently mutated gene was the RAI1 gene, with six synonymous mutations and eight non-frameshift mutations occurring in all seven cases. However, in the COSMIC database, RAI1 it not a known cancer-related gene. A previous study reported that heterozygous mutations located in the chromosomal region 17p11.2 of RAI1 cause Smith-Magenis Syndrome (SMS), which is a rare disorder of analgesia and abnormal body clock.25 Whether RAI1 mutation is related to cardiac myxoma remains to be determined in future studies. Other genes with high mutation rates among the seven cases were TBP (6/7), AR (5/7), AKAP9 (4/7), GIGYF2 (4/7) and PRKAR1A (4/7). The AR and TBP mutations were non-frameshift mutations. The AR is a known cancer gene in the COSMIC database, and mutation of AR can lead to a variety of androgen-sensitive diseases. AR mutations has been shown to be closely related to prostate cancer,26 but no association with myxoma has been reported. AKAP9 is a putative cancer-related gene in the COSMIC database, but there is currently no evidence that it is associated with myxomatosis. In the COSMIC database, GIGYF2 is not a known cancer gene, and the missense mutation rate is 51.98%. In the present study, only one missense mutation of GIGYF2 was identified with no clear relation to myxoma. DCTN1, GALC and GALNS also are not known cancer genes in the COSMIC database. In contrast, PRKAR1A is a known cancer gene and also an established gene distributed in myxoma.9 The finding in our study that mutation of only PRKAR1A could be related to the occurrence of sporadic cardiac myxomas in four cases is of great significance.
The PRKAR1A gene is a tumor suppressor gene that encodes a PKA regulatory 1α subunit.27 Previous research has demonstrated that PRKAR1A mutation is the cause of CNC in approximately 70% cases and localized on Chr 17 q22-24.6,28 However, whether PRKAR1A mutation is involved in the pathogenesis of sporadic cardiac myxoma in the absence of a family history or CNC remains a controversial topic. Early studies found no relationship between PRKAR1A mutation and sporadic cardiac myxoma morbidity. Fogt et al29 analyzed the expression of multiple markers (PRKAR1 9CA, D2S2153, D2S2251 and D2S123) in sporadic cardiac myxoma specimens from 13 patients and found that none showed definite changes in the expression levels of the markers. As a result, they concluded that sporadic cardiac myxomas are not related genetically to CNC. Mantovani et al30 investigated the presence of inactivating mutations in PRKAR1A in cases of sporadically occurring cardiac myxomas in 29 patients and did not find any mutations by direct sequencing.
Recent data suggest that the loss of PRKAR1A protein expression may play a role in isolated myxoma tumorigenesis. Maleszewski et al31 analyzed the immunohistochemical staining results for 110 cardiac myxoma tissue specimens and observed the absence of PRKAR1A antigenicity in all seven cases with CNC and that 32% of isolated cardiac myxomas were similarly nonreactive. PRKAR1A gene sequencing analysis in their study further confirmed that 67% of CNC myxomas and 31% of isolated myxomas had pathogenic PRKAR1A mutations. Roque et al18 presented the case of a 48-year-old Caucasian woman who developed progressive brain metastases one year after removal of a proven sporadic atrial myxoma. Genetic analysis of this case showed multiple mutations within the PRKAR1A gene in tissue samples from both the brain metastasis and cardiac tumor, which provides further evidence that PRKAR1A mutation can occur in sporadic cardiac myxomas. In the present study, targeted NGS analysis of cardiac myxoma specimens from seven sporadic cases identified five pathogenic mutations of PRKAR1A distributed in four sporadic cardiac myxoma cases, for a PRKAR1A inactivating mutation rate of 57.2% (4/7). From these results, we conclude that mutation of the PRKAR1A gene is one pathogenic mechanism of sporadic cardiac myxoma.
To further explore the pathogenic role of PRKAR1A mutation in sporadic cardiac myxoma, we examined the PRKAR1A gene mutations via SNV and INDEL variant analysis. The six PRKAR1A mutations found in the four cases included one synonymous mutation (c.678C > T), one nonsense mutation (c.366C > G), one missense mutation (c.496C > T), and three frameshift mutations (c.61_62insAC, c.273_274insAAAG and c.569delG). Yinet al32 reported that mice lacking PRKAR1A protein specifically in cardiomyocytes ultimately develop heart failure and myxoma-like phenotype. The majority of PRKAR1A defects are premature stop codons generated by nonsense, missense or frameshift (insertions, deletions or splice-site modifications) mutations.33 Additionally, the degradation of mutant mRNAs via nonsense-mediated decay was shown to be the cause of PRKAR1A haploinsufficiency,34 which then activates cAMP signaling and promotes downstream cellular processes that lead to tumorigenesis.35 Therefore, the present study demonstrates the occurrence of PRKAR1A mutation in myxoma specimens of four sporadic cardiac myxoma cases without CNC or a family history of cardiac myxoma. All of the identified pathogenic mutationsof PRKAR1A,which include nonsense, missense and frameshift mutations, lead to premature appearance of terminators (Table 2, Figure 2).
Inactivating germline mutations of the PRKAR1A gene have been reported in the literature in a majority of patients with CNC.36 In contrast to the genetic abnormalities observed in CNC patients, few single gene mutations leading to sporadic cardiac myxoma have been found to date.16,18 Moreover, the genetic basis of sporadic cardiac myxoma has yet to be fully elucidated. In the present genetic study, we used both cardiac myxoma specimens and matched peripheral blood samples from the same patients for Sanger sequencing analysis. A major discovery of the present study is that all the pathogenic mutations (missense, nonsense, and frameshift mutations) of PRKAR1A were found only in the cardiac myxoma samples, with no pathogenic mutations found in the matched peripheral blood samples, except for one synonymous mutation(c.678C > T). Therefore, somatic mutations in the PRKAR1A gene may be causative mutations for sporadic cardiac myxoma.
In conclusion, our study demonstrated that mutation of the PRKAR1A gene was associated with the pathogenesis of sporadic cardiac myxoma in four cases lacking a family history of cardiac myxoma or CNC. Notably, the PRKAR1A mutations identified in the cardiac myxoma specimens were somatic mutations, which were acquired and non-inheritable.
Conclusion
Pathogenic mutations of the PRKAR1A gene were identified in tumor specimens from four cases of sporadic cardiac myxoma, and the absence of these mutations in peripheral blood samples demonstrated that they were somatic mutations.
Supplementary Files
Supplementary file 1 contains Tables S1-S3.
(pdf)
Acknowledgements
The authors appreciate the contributions of all members of the Department of Cardiac Surgery as well as the anesthetists. We also acknowledge the company workers that provided genetic testing.
Competing Interests
The authors declare that they have no conflict of interest.
Data Availability Statement
The datasets generated and analyzed during the current study are not publicly available because none of the data types require uploading to a public repository; however, the data are available from the corresponding author on reasonable request.
Ethical Approval
This work was supported by a grant from the National Natural Science Foundation of China (#81600150) to J.-N. S.
Funding
This work was supported by a grant from Science and Technology Development Plan Project of Jilin Province, China (#20210101332JC) to Yunpeng Sun and the National Natural Science Foundation of China (#81600150) to Jingnan Sun.
References
- Jain S, Maleszewski JJ, Stephenson CR, Klarich KW. Current diagnosis and management of cardiac myxomas. Expert Rev Cardiovasc Ther 2015; 13(4):369-75. doi: 10.1586/14779072.2015.1024108 [Crossref] [ Google Scholar]
- Vaideeswar P, Butany JW. Benign cardiac tumors of the pluripotent mesenchyme. Semin Diagn Pathol 2008; 25(1):20-8. doi: 10.1053/j.semdp.2007.10.005 [Crossref] [ Google Scholar]
- Nehaj F, Sokol J, Mokan M, Jankovicova V, Kovar F, Kubaskova M. Outcomes of patients with newly diagnosed cardiac myxoma: a retrospective multicentric study. Biomed Res Int 2018; 2018:8320793. doi: 10.1155/2018/8320793 [Crossref] [ Google Scholar]
- Samanidis G, Khoury M, Balanika M, Perrea DN. Current challenges in the diagnosis and treatment of cardiac myxoma. Kardiol Pol 2020; 78(4):269-77. doi: 10.33963/kp.15254 [Crossref] [ Google Scholar]
- Elbardissi AW, Dearani JA, Daly RC, Mullany CJ, Orszulak TA, Puga FJ. Survival after resection of primary cardiac tumors: a 48-year experience. Circulation 2008; 118(14 Suppl):S7-15. doi: 10.1161/circulationaha.107.783126 [Crossref] [ Google Scholar]
- Wilkes D, McDermott DA, Basson CT. Clinical phenotypes and molecular genetic mechanisms of Carney complex. Lancet Oncol 2005; 6(7):501-8. doi: 10.1016/s1470-2045(05)70244-8 [Crossref] [ Google Scholar]
- Yang M, Long B, Xu J, Yu J, Li X, Ye F. Clinical manifestations and molecular biology of one case of Carney complex: a case report. Iran J Public Health 2018; 47(4):597-602. [ Google Scholar]
- Tirosh A, Hamimi A, Faucz F, Aharon-Hananel G, Zavras PD, Bonella B. Liver findings in patients with Carney complex, germline PRKAR1A pathogenic variants, and link to cardiac myxomas. Endocr Relat Cancer 2020; 27(6):355-60. doi: 10.1530/erc-19-0517 [Crossref] [ Google Scholar]
- Burke A, Tavora F. The 2015 WHO classification of tumors of the heart and pericardium. J Thorac Oncol 2016; 11(4):441-52. doi: 10.1016/j.jtho.2015.11.009 [Crossref] [ Google Scholar]
- Correa R, Salpea P, Stratakis CA. Carney complex: an update. Eur J Endocrinol 2015; 173(4):M85-97. doi: 10.1530/eje-15-0209 [Crossref] [ Google Scholar]
- Pitsava G, Zhu C, Sundaram R, Mills JL, Stratakis CA. Predicting the risk of cardiac myxoma in Carney complex. Genet Med 2021; 23(1):80-5. doi: 10.1038/s41436-020-00956-3 [Crossref] [ Google Scholar]
- Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 1985; 64(4):270-83. doi: 10.1097/00005792-198507000-00007 [Crossref] [ Google Scholar]
- Feng J. Timely screening for Carney complex and PRKAR1A gene mutations. J Thorac Cardiovasc Surg 2016; 152(5):1440-1. doi: 10.1016/j.jtcvs.2016.07.060 [Crossref] [ Google Scholar]
- Kamilaris CDC, Faucz FR, Voutetakis A, Stratakis CA. Carney complex. Exp Clin Endocrinol Diabetes 2019; 127(2-03):156-64. doi: 10.1055/a-0753-4943 [Crossref] [ Google Scholar]
- Sun Y, Chen X, Sun J, Wen X, Liu X, Zhang Y. A novel inherited mutation in PRKAR1A abrogates preRNA splicing in a Carney complex family. Can J Cardiol 2015; 31(11):1393-401. doi: 10.1016/j.cjca.2015.05.018 [Crossref] [ Google Scholar]
- Gošev I, Paić F, Durić Z, Gošev M, Ivčević S, Jakuš FB. Cardiac myxoma the great imitators: comprehensive histopathological and molecular approach. Int J Cardiol 2013; 164(1):7-20. doi: 10.1016/j.ijcard.2011.12.052 [Crossref] [ Google Scholar]
- Buttar R, Hoefen R, Funderburk M, Fallone E, Baibhav B. Sporadic form of recurrent atrial myxoma: the blob strikes back. Cureus 2020; 12(8):e9745. doi: 10.7759/cureus.9745 [Crossref] [ Google Scholar]
- Roque A, Kimbrough T, Traner C, Baehring JM, Huttner A, Adams J. Somatic PRKAR1A mutation in sporadic atrial myxoma with cerebral parenchymal metastases: a case report. J Med Case Rep 2019; 13(1):389. doi: 10.1186/s13256-019-2317-z [Crossref] [ Google Scholar]
- Wang HY, Zhang XB, Zheng JJ, Deng Y, Wang YL, Song YX, et al. [Clinicopathologic features of cardiac myxoma--a report of 47 cases]. Ai Zheng 2006;25(7):892-5. [Chinese].
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25(14):1754-60. doi: 10.1093/bioinformatics/btp324 [Crossref] [ Google Scholar]
- Jin ZB, Huang XF, Lv JN, Xiang L, Li DQ, Chen J. SLC7A14 linked to autosomal recessive retinitis pigmentosa. Nat Commun 2014; 5:3517. doi: 10.1038/ncomms4517 [Crossref] [ Google Scholar]
- Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017; 19(1):4-23. doi: 10.1016/j.jmoldx.2016.10.002 [Crossref] [ Google Scholar]
- Yokoyama S, Nagao K, Higashida A, Aoki M, Yamashita S, Fukuda N. Diagnosis of Carney complex following multiple recurrent cardiac myxomas. Gen Thorac Cardiovasc Surg 2022; 70(1):87-91. doi: 10.1007/s11748-021-01719-w [Crossref] [ Google Scholar]
- Siordia JA. Medical and surgical management of Carney complex. J Card Surg 2015; 30(7):560-7. doi: 10.1111/jocs.12575 [Crossref] [ Google Scholar]
- Carmona-Mora P, Canales CP, Cao L, Perez IC, Srivastava AK, Young JI. RAI1 transcription factor activity is impaired in mutants associated with Smith-Magenis syndrome. PLoS One 2012; 7(9):e45155. doi: 10.1371/journal.pone.0045155 [Crossref] [ Google Scholar]
- Li Q, Deng Q, Chao HP, Liu X, Lu Y, Lin K. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat Commun 2018; 9(1):3600. doi: 10.1038/s41467-018-06067-7 [Crossref] [ Google Scholar]
- Massobrio L, Nasti S, Martinuzzi C, Chiarella F, Montecucco F, Rosa GM. Mutation analysis of PRKAR1A gene in a patient with atrial myxoma. Clin Lab 2016; 62(4):731-4. doi: 10.7754/clin.lab.2015.150838 [Crossref] [ Google Scholar]
- Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L. Mutations in regulatory subunit type 1A of cyclic adenosine 5’-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 2009; 94(6):2085-91. doi: 10.1210/jc.2008-2333 [Crossref] [ Google Scholar]
- Fogt F, Zimmerman RL, Hartmann CJ, Brown CA, Narula N. Genetic alterations of Carney complex are not present in sporadic cardiac myxomas. Int J Mol Med 2002; 9(1):59-60. [ Google Scholar]
- Mantovani G, Bondioni S, Corbetta S, Menicanti L, Rubino B, Peverelli E. Analysis of GNAS1 and PRKAR1A gene mutations in human cardiac myxomas not associated with multiple endocrine disorders. J Endocrinol Invest 2009; 32(6):501-4. doi: 10.1007/bf03346496 [Crossref] [ Google Scholar]
- Maleszewski JJ, Larsen BT, Kip NS, Castonguay MC, Edwards WD, Carney JA. PRKAR1A in the development of cardiac myxoma: a study of 110 cases including isolated and syndromic tumors. Am J Surg Pathol 2014; 38(8):1079-87. doi: 10.1097/pas.0000000000000202 [Crossref] [ Google Scholar]
- Yin Z, Jones GN, Towns WH 2nd, Zhang X, Abel ED, Binkley PF. Heart-specific ablation of PRKAR1A causes failure of heart development and myxomagenesis. Circulation 2008; 117(11):1414-22. doi: 10.1161/circulationaha.107.759233 [Crossref] [ Google Scholar]
- Di Vito A, Mignogna C, Donato G. The mysterious pathways of cardiac myxomas: a review of histogenesis, pathogenesis and pathology. Histopathology 2015; 66(3):321-32. doi: 10.1111/his.12531 [Crossref] [ Google Scholar]
- Veugelers M, Wilkes D, Burton K, McDermott DA, Song Y, Goldstein MM. Comparative PRKAR1A genotype-phenotype analyses in humans with Carney complex and PRKAR1A haploinsufficient mice. Proc Natl Acad Sci U S A 2004; 101(39):14222-7. doi: 10.1073/pnas.0405535101 [Crossref] [ Google Scholar]
- Kondo K, Harada M, Konomoto T, Hatanaka M, Nunoi H. Novel PRKAR1A mutation in Carney complex with cardiac myxoma. Pediatr Int 2017; 59(7):840-1. doi: 10.1111/ped.13302 [Crossref] [ Google Scholar]
- Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet 2000; 26(1):89-92. doi: 10.1038/79238 [Crossref] [ Google Scholar]